






生信碱移
7类单细胞算法
单细胞技术打破了传统生物学研究中模糊的视角,使研究者能够单独研究每个细胞的基因或其他特征,而不是观察一群细胞的平均行为。然而,这项技术也带来了挑战:数据的收集和分析成本高昂,研究者通常需要在分辨率、通量和组织位置之间权衡;尽管在单细胞生物学中,研究者可以深入了解单个细胞的信息,但有时确难以精准确定其来源。
▲ HCA 项目负责人 Aviv Regev 教授:美国国家科学院院士、Broad研究所教授、单细胞基因组和基因调控回路的计算和系统生物学的先驱。
作为单细胞技术设计和应用的前沿项目,Human Cell Atlas(HCA)旨在对人类的每种细胞类型进行全面的分类。该项目于 2016 年启动,目前已对数亿个单细胞进行了剖析,产生了约 440 篇研究论文,并催生了几十种计算软件。
就在 2024 年 11 月 20 日,Nature [IF: 50.5] 在 "Computational technologies of the Human Cell Atlas" 中重点介绍了 HCA 的七种核心技术工具。这些工具包括:用于细胞分类和数据搜索的平台、低成本获取空间或多模态数据的快捷方法,以及描述细胞交互与病变细胞如何响应治疗的模型。
一、细胞搜索与注释工具
单细胞测序生成的数据量庞大且复杂,如何对这些数据进行高效标注、分类和整合,成为研究人员亟待解决的难题。
为了应对这一挑战,研究人员开发了多种自动标注工具,其中之一便是PopV(Popular Vote)。PopV 的设计灵感源自“投票”机制——它整合了八种不同的细胞标注算法,并对每个细胞的类型进行投票。所有算法一致认定某种细胞类型时,结果被认为是高度可靠的;而如果存在分歧,研究人员可以通过“不确定性评分”来衡量结果的可信度。这样的多算法投票机制显著提高了标注的效率和准确性。
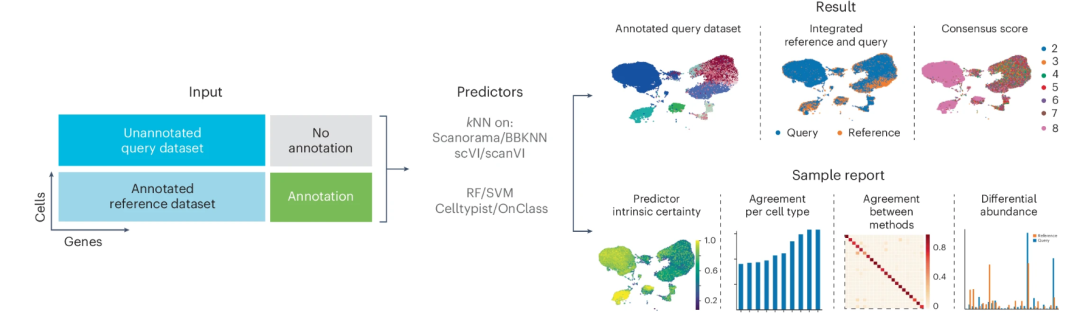
▲ PopV 将未标注的查询数据集和已标注的参考数据集作为输入。每种专家算法都会预测查询数据集上的标签,从而得出细胞类型注释。通过对这些方法的一致性进行评分,可以量化各自标签转移的确定性。DOI: 10.1038/s41588-024-01993-3。
PopV的开发者使用 Tabula Sapiens 数据集对其进行训练,该数据集涵盖来自 15 个个体的 24 个器官、近 50 万个细胞。PopV 在 Human Lung Cell Atlas (人类肺细胞图谱,包含超过2300万个细胞)中的测试中表现卓越,其预测与人工标注结果高度一致,准确性达到了92%,比单一算法平均准确率提高了15%。
二、细胞相似性工具
一旦研究人员发现了一种有趣的细胞类型或状态,他们可能会想知道它还会在哪里出现。为此,Regev 和她的同事开发了SCimilarity 来回答这个问题。该软件可以帮助研究者识别与目标细胞类型相似的细胞群体,就像测序分析中使用 BLAST 算法寻找相关的参考序列一样。
简单来说,每个细胞最初都是由约 2 万个人类基因的表达来定义的,但 SCimilarity 将这些基因压缩成 128 个细胞身份的关键特征;而匹配算法使用这些特征。开发团队利用了来自近 400 个数据集的 2300 多万个细胞,只需几秒钟就能完成细胞搜索。
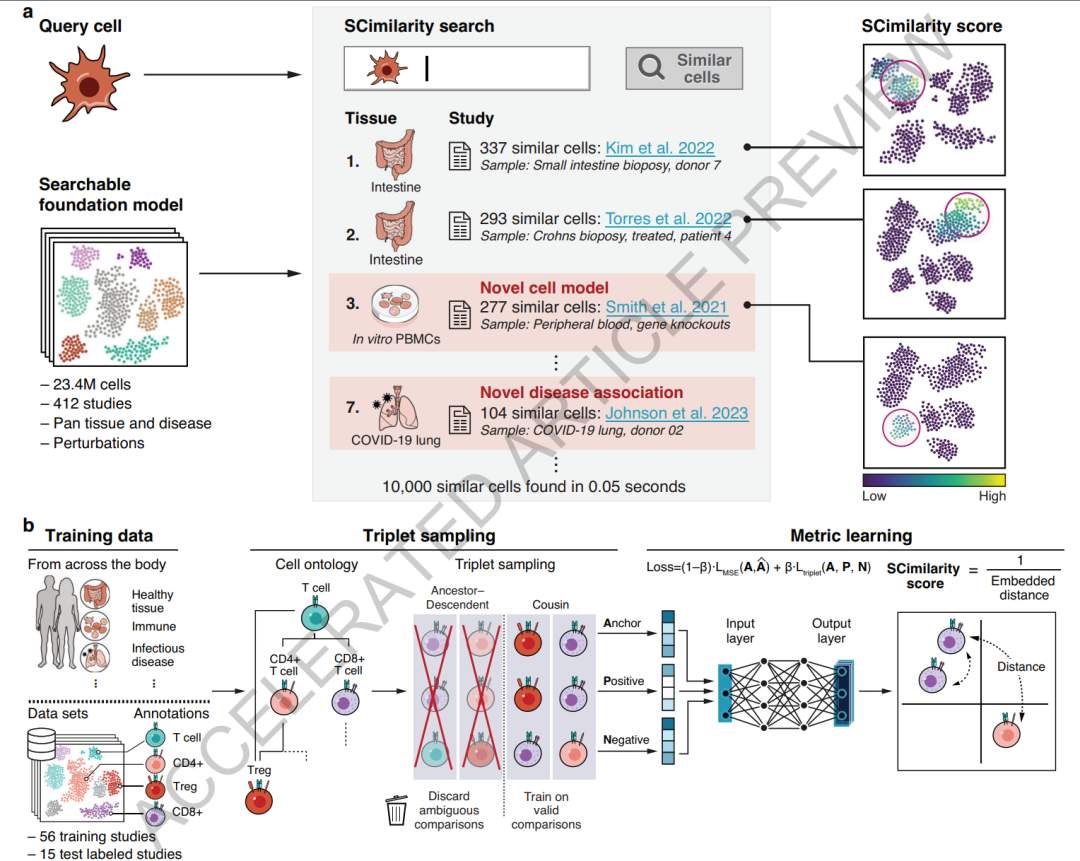
▲ SCimilarity 是一种基于度量学习的细胞搜索工具,能够在包含 23.4M 细胞轮廓的参考数据库中高效定位与查询细胞相似的样本。它通过三元组损失(Triplet Loss)训练神经网络,利用细胞本体论注释,从全身组织中选择锚点细胞、相似细胞和不相似细胞进行学习,从而实现对细胞相似性关系的高精度建模,形成基础模型并支持大规模细胞图谱的精确搜索与分析。DOI: 10.1038/s41586-024-08411-y。
研究团队在 17 项体外(实验室培养)和体内(活体)研究中,分析了约 42000 个细胞。在这些数据中,他们意外地在一种三维水凝胶系统中培养的白细胞中,发现了一种能制造造血干细胞的细胞。这种发现是通过 SCimilarity 工具进行的大规模数据匹配和分析得出的。研究团队进一步在实验室中重新培育这些细胞,并证实它们与纤维化肺组织中的细胞具有相似性。这种联系在研究初期并不显而易见,但 SCimilarity 工具 通过分析数据间的深层相似性,揭示了这一意外发现。
三、高效数据推断
高分辨率或高通量单细胞实验的费用让许多研究小组望而却步。但研究者们正在开发变通方法,利用人工智能和机器学习从更小或更简单的数据集推断单细胞或空间数据。
其中一个例子是 scSemiProfiler。假设研究人员需要单细胞 RNA 图谱,但只能负担大量 RNA 测序的费用。为了帮助他们充分利用资源,scSemiProfiler 利用批量数据和生成式人工智能生成单细胞图谱的可能分布。麦吉尔大学健康中心的计算生物学家丁俊(Jun Ding)说,这就像拍摄一张低分辨率的数码照片,然后推断出高分辨率的照片。
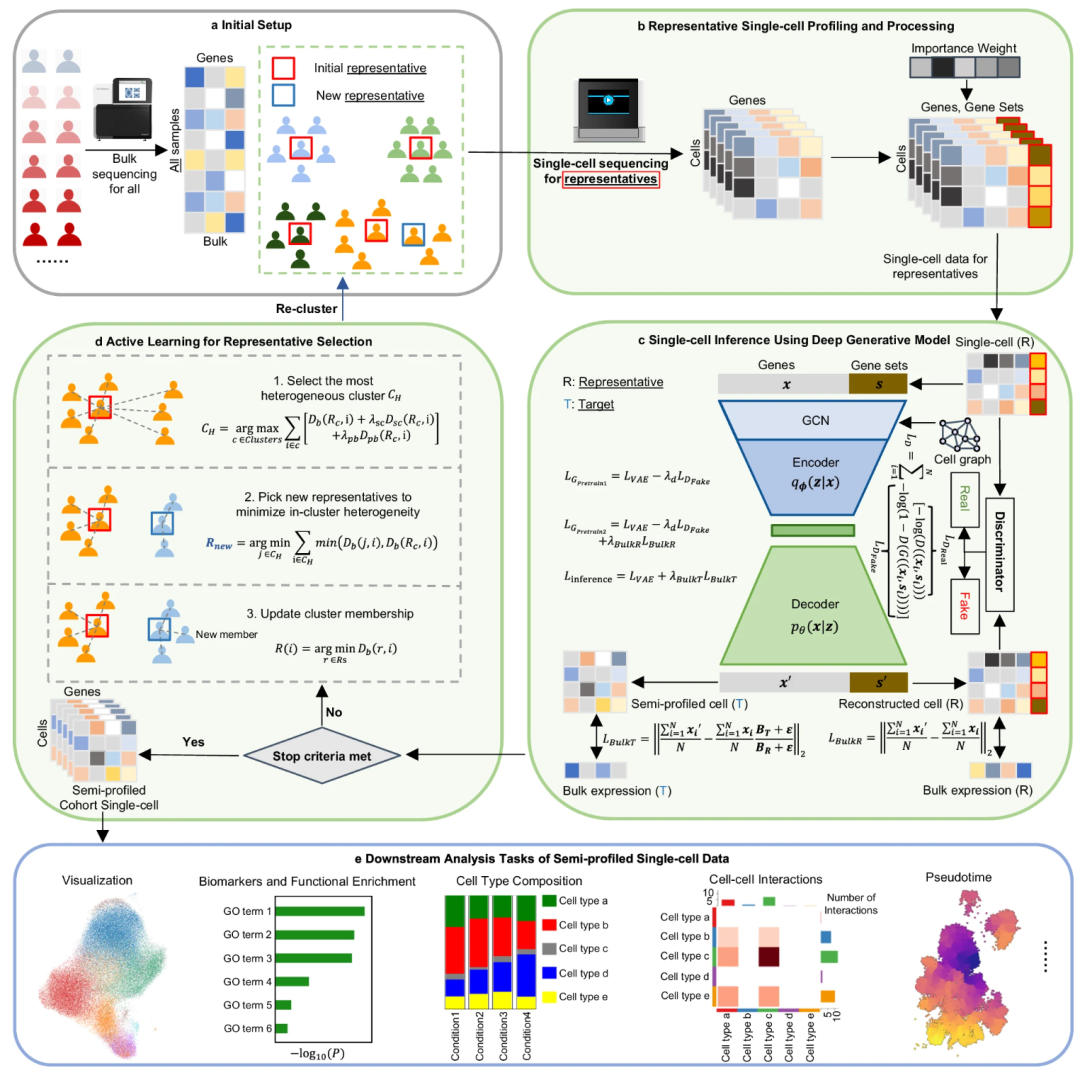
▲ scSemiProfiler 的核心原理是利用生成式人工智能模型,将 bulk RNA 测序数据转化为单细胞RNA测序数据的高分辨率分布。首先,通过少量实际的单细胞测序数据作为初始输入,模型学习代表性细胞类型的特征分布,然后结合 bulk 数据进行推断,生成其他样本的单细胞分布。模型还可通过主动学习算法优化采样策略,建议额外的单细胞测序需求,从而显著降低实验成本,同时提供接近真实的单细胞数据,为后续分析奠定基础。
Ding 及其同事对来自 124 名 COVID-19 患者和非 COVID-19 患者免疫细胞的单细胞 RNA 图谱进行了 scSemiProfiler 测试。该程序能够根据每个样本的 bulk 测序和具有代表性的子集(原始样本中仅有 28 个样本)的单细胞序列生成正确的单细胞图谱。研究人员估计,这种方法能为研究人员的类似研究节省近 12.5 万美元,因为它能将所需的单细胞测序减少约 80%。
四、空间转录组推断
同样,Regev 和她的同事们正在利用机器学习作为一种捷径,从现成的资源中生成空间分辨的单细胞 RNA 测序数据:用血红素和伊红(H&E)染色的组织切片。这种染色技术已经使用了一个多世纪,实验室和医院的档案室里堆满了这些切片。由于这些染色模式在某种程度反应基因表达等分子特征,Regev 和她的团队想知道他们是否能利用 H&E 信息生成目前“花里胡哨”的数据:空间转录组数据。
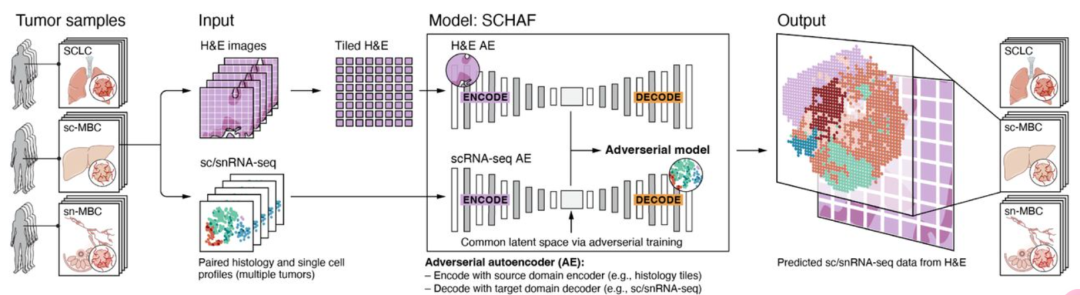
▲ SCHAF 利用组织切片(H&E 染色)图像预测单细胞组学数据。通过对组织图像进行标准化和分块,SCHAF 使用图像自动编码器将图像编码到潜在空间,并通过对抗性训练使其与基因表达编码器的潜在空间一致。在推断阶段,SCHAF 接收输入的组织图像,生成与特定肿瘤类型对应的空间位置上的单细胞基因表达分布,实现高效的组织组学数据推断。DOI: 10.1101/2023.03.21.533680。
事实上,研究人员可以做到这一点。他们的程序 SCHAF (组织学单细胞组学分析框架)有两个版本。配对版本使用来自同一切片组织的 H&E 染色和有限的空间转录组学数据,以及来自相邻切片的单细胞 RNA 图谱进行训练。相比之下,非配对 SCHAF 是在没有任何空间 RNA 数据的情况下进行训练的。研发人员表示:"非配对条件下仍然能得到一个很好的模型,但可能没有那么强大。研究人员在已匹配 H&E、转录组和空间 RNA 数据的数据集上测试了 SCHAF,其中两个数据集是乳腺癌数据集,一个是小细胞肺癌数据集。
五、多模态数据整合与预测
除了像 SCHAF 那样使用一种类型的数据来预测另一种类型外,计算机模型还可以将来自同一样本的多种类型数据纳入其中。这就是 multiDGD 方法的目标,它利用来自同一细胞的 RNA 表达和染色质可及性数据建立生物学模型。
multiDGD 的输入基于约 2 万个人类基因的表达水平以及整个基因组数十万个区段的染色质状态(开放或封闭)--每个细胞约有 20 万个特征。这些因素被简化为一组具有代表性的 20 个左右的特征,然后输入模型。
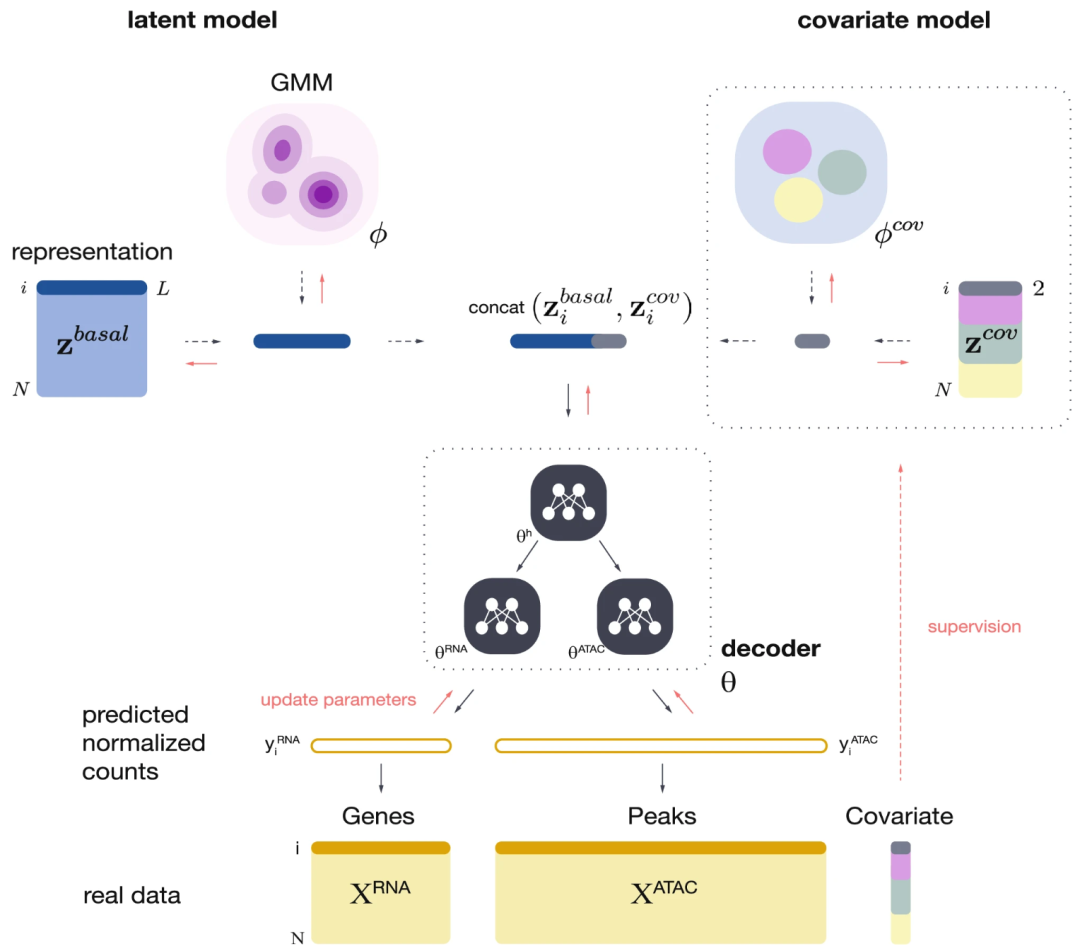
▲ multiDGD 是一种多组学深度生成模型,结合无监督和监督方法,通过高斯混合模型(GMM)建模潜在空间,同时引入协变量信息以增强解释力。模型利用潜在表示生成特定模态(如 RNA 和 ATAC)的归一化均值,并结合样本计数深度预测组学数据密度。通过反向传播优化参数,multiDGD 实现了对多组学数据的高效建模与生成。DOI: 10.1038/s41467-024-53340-z。
multiDGD的优势在于其对多模态数据的整合能力。单一的数据类型往往无法全面反映细胞状态,例如,基因表达数据可以揭示细胞的功能状态,而染色质开放状态则反映基因的可及性和潜在的转录活性。通过结合这些数据,multiDGD 可以为研究人员提供一个更为全面的细胞动态变化图景。例如,在干细胞分化的研究中,multiDGD 揭示了基因表达和染色质状态的协同变化,从而为理解细胞命运决策提供了新的视角。
六、细胞交互模拟
研究人员还可以对模型提出问题,比如说扰乱一个基因或放大一个基因的表达。在一个例子中,研究小组测试了沉默 41 个转录因子在 silico 中如何改变目标基因的染色质可及性。一个名为 CellAgentChat 的模型可以推断出一定距离范围内的细胞间相互作用,该模型将每个细胞视为一个自主体(agent),模拟其在复杂环境中的行为。每个细胞自主体具备数字“受体”,可以接收其他细胞释放的分子信号,并基于这些信号激活相应的基因表达模式。
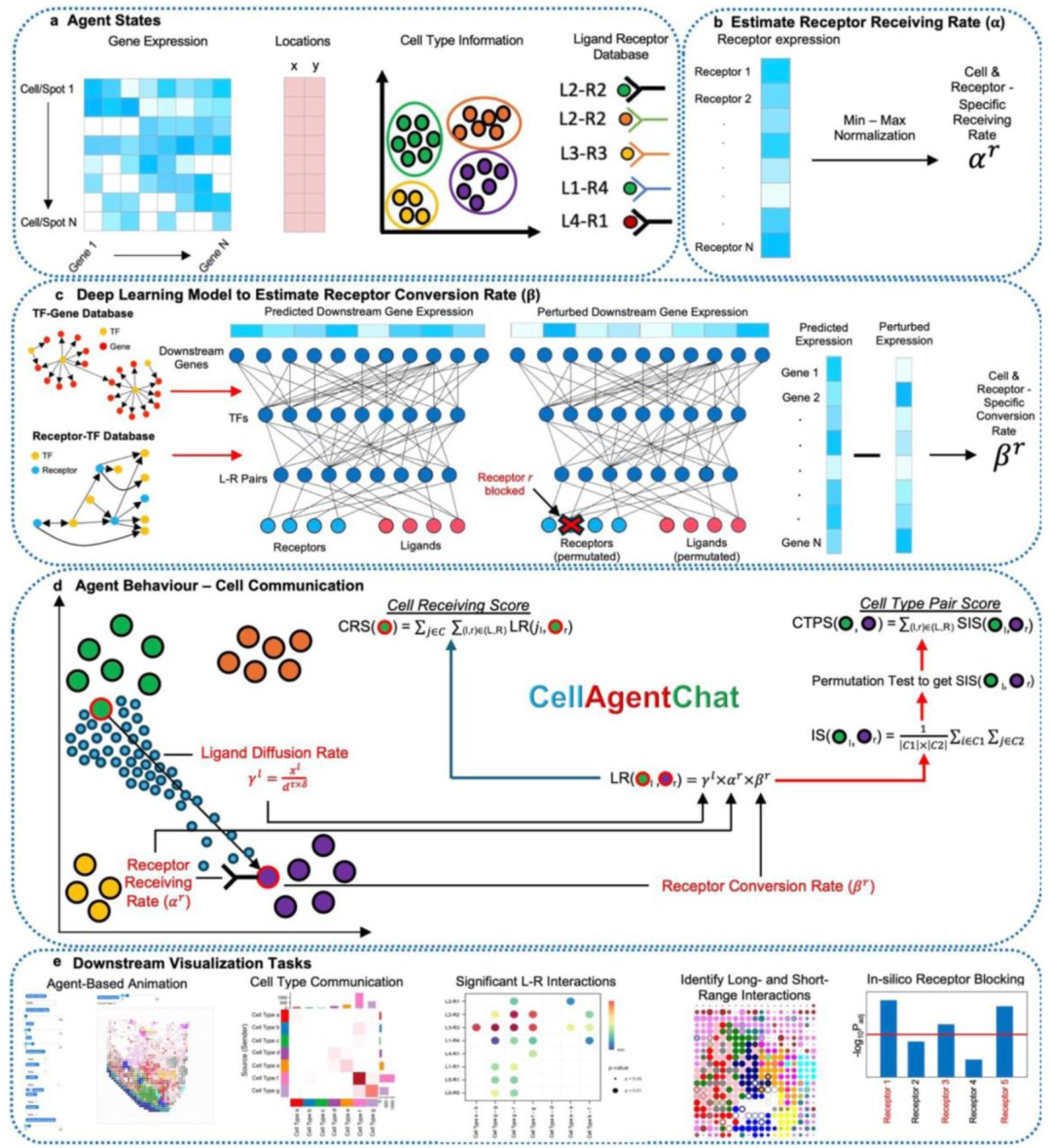
▲ CellAgentChat 是一种基于细胞代理的通信建模工具,结合单细胞转录组数据、配体-受体数据库(默认 CellTalkDB)及可选的空间坐标,模拟和分析细胞间通信。其核心包括:通过深度学习模型结合转录因子与基因交互知识,计算配体扩散率、受体接收率及转化率,生成配体-受体得分;同时支持长短距离交互的优先级控制和受体阻断的模拟分析,以评估下游基因的扰动。工具可实现细胞间通信的可视化、显著配体-受体对的识别、长短距离交互特征分析,以及用于药物发现的模拟实验。DOI: 10.1101/2023.08.23.554489。
通过使用 CellAgentChat,研究人员可以在实验前模拟不同药物对细胞间信号的影响,从而优化药物筛选过程。研究小组在乳腺癌数据集进行了这种尝试,结果证实表皮生长因子受体(一种已知的致病因子和药物靶点)也是其交互作用中的关键交互因子。
七、疾病进程与药物筛选
研究小组还开发了一个名为 UNAGI 的模型,专门用于药物测试,重点研究细胞如何随时间变化。UNAGI 模型通过生成式神经网络,模拟疾病进展并筛选潜在药物,为肺纤维化等疾病的治疗提供了新见解。
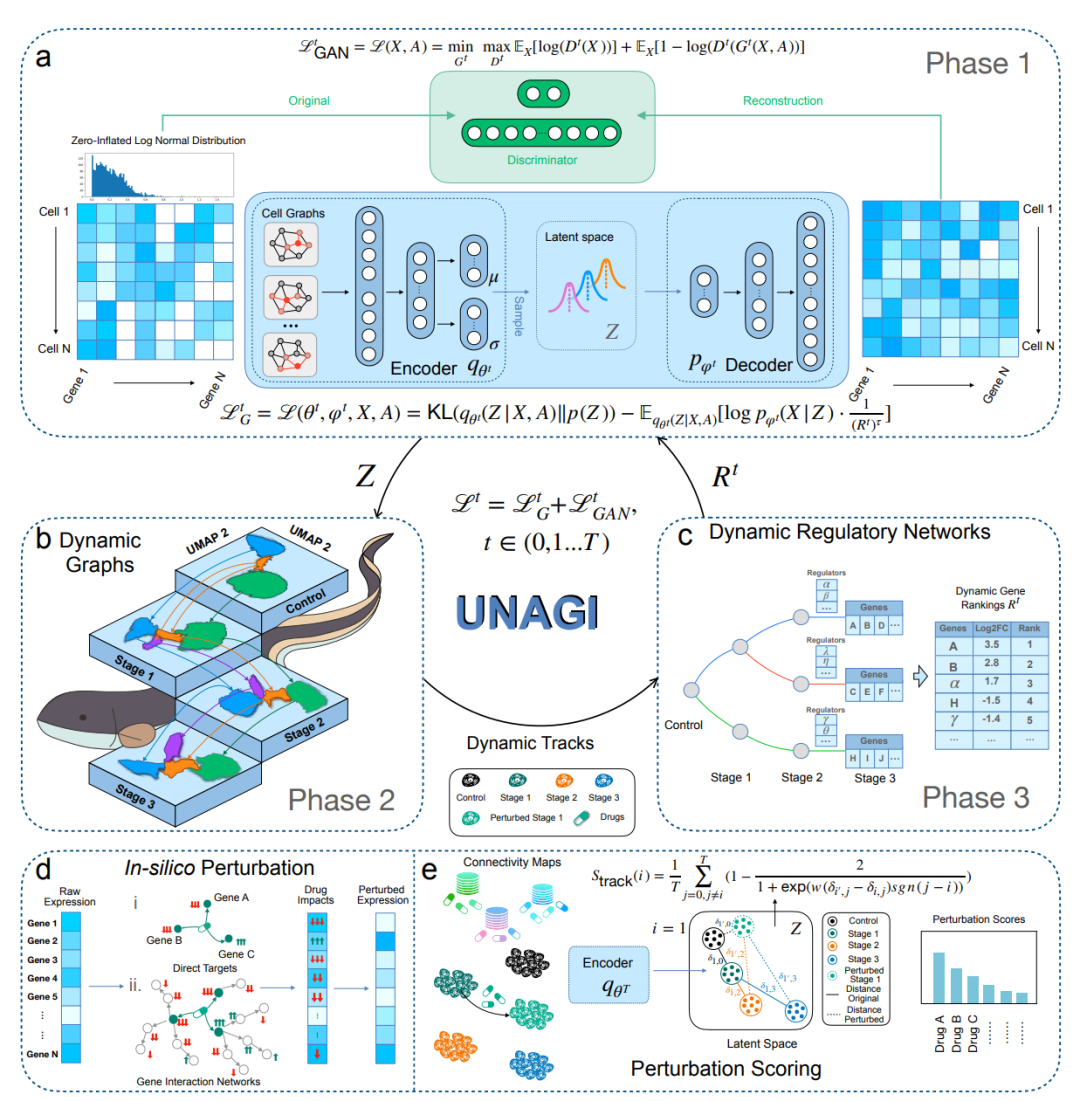
▲ UNAGI 是一款结合 VAE-GAN 和图卷积网络的工具,用于解析复杂疾病的细胞动力学与潜在治疗药物。通过生成潜在空间嵌入 ('Z'),UNAGI 构建时间动力学图,识别关键基因调控因子,并通过模拟药物干预评估其对疾病细胞的影响。模型通过在体内外的基因表达调整,识别潜在药物候选。DOI: 10.21203/rs.3.rs-3676579/v1。
具体来讲,研究小组将特发性肺纤维化四个阶段的肺病数据输入 UNAGI,创建了一个虚拟的疾病进展 "沙盘",每个细胞都由深度生成神经网络中的几十个特征表示。利用这个模型,研究人员可以推断出基因表达随着疾病的进展会发生怎样的变化,并测试不同的药物是否会将细胞推回到早期的基因表达谱,或者推向更健康的基因表达谱。在研究人员的筛选中出现了一种已获美国食品药品管理局批准的药物:宁替达尼,这是一种生长因子抑制剂,可防止成纤维细胞的繁殖。
牛导就是不一样
方法学层面都不难实现
主要还是生成模型VAE这些
数据和计算资源才是重点
能被 Nature 单独开一节报道也够猛
欢迎各位老铁关注

















 1191
1191

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









