










在过去的几年里,我们已经见识到单细胞转录组学技术在生物学与医学研究领域取得了令人瞩目的进展。它所带来的主要突破在于能够针对单个细胞进行基因表达谱的测量,从而揭示不同细胞类型及其亚群间的异质性。但是,单细胞间的异质性可能存在于基因组、表观基因组、转录组、蛋白质和代谢等各个层面。
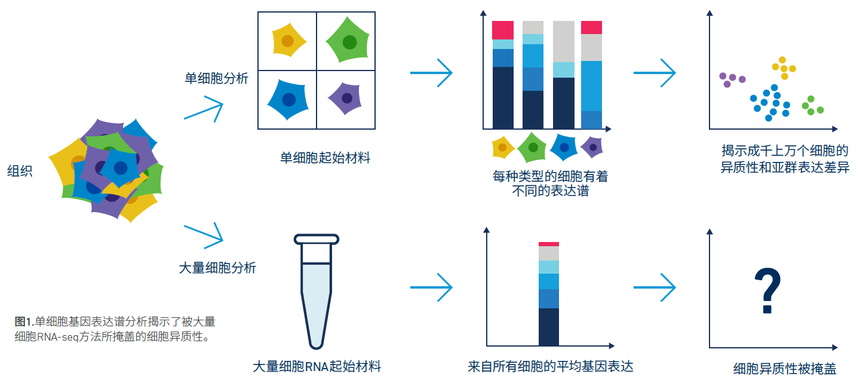
单细胞多组学为深入探索这些不同层面上的异质性提供了前所未有的机会。整合基因组、表观基因组、转录组、蛋白质组以及代谢组等多种信息,单细胞多组学为阐明细胞功能与疾病机制奠定了更加全面的基础。借助多组学数据的综合分析,研究者不仅能捕捉到细胞状态的动态变化,还可精确鉴别关键调控网络和通路。据此,我们能够更好地理解不同细胞在发育、生理以及病理过程中的特殊作用,从而为精准医学、药物研发以及个性化治疗策略提供重要支撑。
回顾近十年的单细胞文章,我们可以看到单细胞转录组和单细胞多组学的发文数量都呈现指数级的增长,单细胞多组学的高分文章(IF>10)甚至在近6年都保持稳步增长。
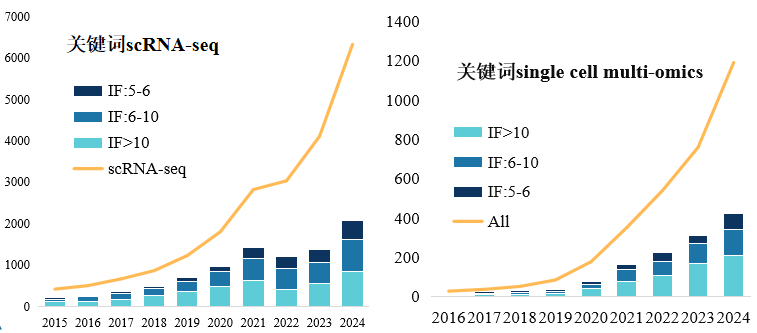
图:Pubmed中单细胞转录组和单细胞多组学发表文章数量
单细胞表观组
染色质可及性、DNA甲基化、组蛋白修饰和染色质构象四种表观特征,虽然在整体细胞群(bulk)层面已被深入研究,但随着技术的突破,现可在单细胞水平检测,从而精准解析功能元件与染色质状态。将单细胞表观组与单细胞转录组相结合,已成为多组学研究和高分单细胞文章的核心方向,众多研究和分析工具正是从这一视角出发。基于这一背景,本文将重点盘点单细胞表观多组学的技术进展与应用实践。
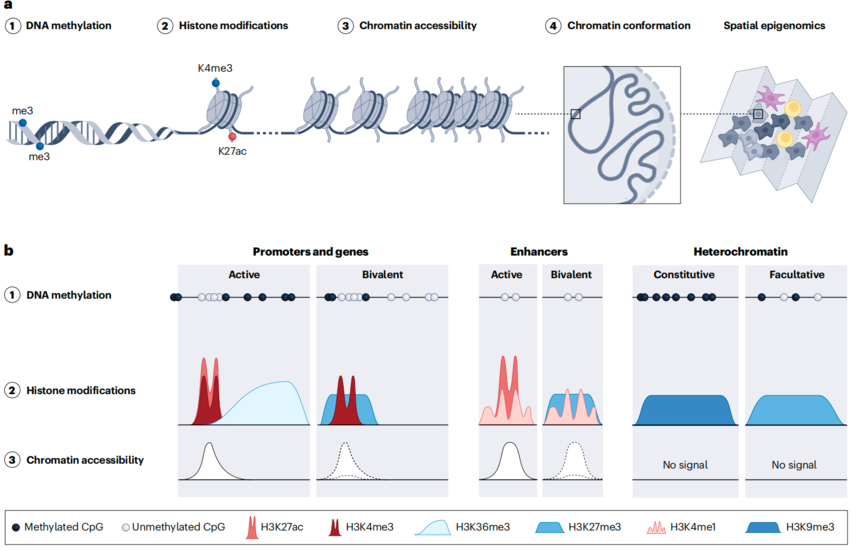
图:单细胞水平可以分析的表观修饰。通过一系列的表观修饰可以表征功能元件和染色质状态[1]。
2025年2月,一篇发表于Nature review cancer的重磅综述探讨了表观异质性作为肿瘤进化的来源,文章回顾了单细胞表观组应用于人类癌症的研究。单细胞表观组在癌症的应用还较为空白,其中应用最广泛的技术只有单细胞ATAC-seq,其次是针对组蛋白修饰检测的单细胞CUT&Tag。我们可以从癌症的研究一窥单细胞表观组的整个研究现状,还有较大的应用潜力。
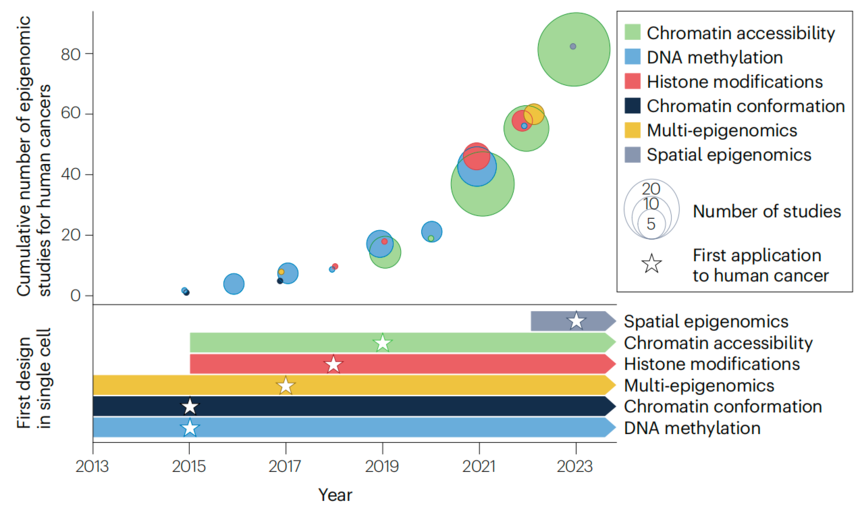
图:单细胞表观组在人类癌症中的应用[1]
单细胞多组学技术概述
以10xGenomics的单细胞转录组为例,主要通过微流控系统将带有条形码的凝胶珠与单细胞悬液或细胞核悬液进行分选,形成油包水的微滴结构。在这个过程中,油包水包裹着凝胶微珠和细胞,然后细胞会被裂解,使细胞内的mRNA自由释放出来。凝胶微珠上的捕获序列可以有效地捕获游离mRNA的polyA尾。接下来,进行逆转录、文库构建、测序和数据分析,从而获得单细胞层面的转录组信息。
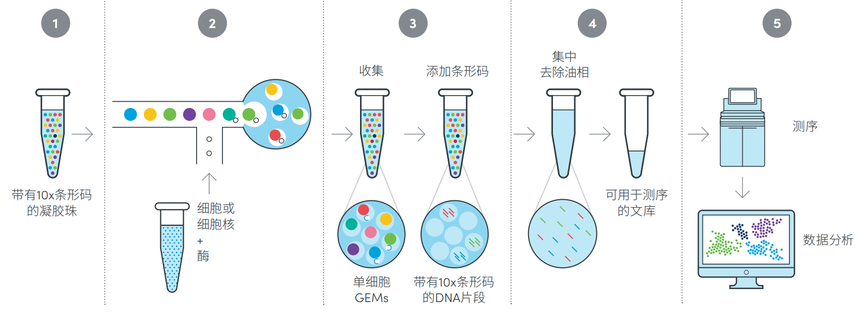
图:10xGenomics单细胞转录组平台技术流程
单细胞3’转录组的每个凝胶微珠(Gel Beads)表面有很多核酸序列(分为四部分:R1、10x Barcode、UMI和Capture Sequence),它最后一段的捕获序列是poly(dT),用于捕获mRNA的poly A尾巴。如果是单细胞ATAC-seq的话,最后的捕获序列则是用来捕获Tn5酶转座后的接头序列。
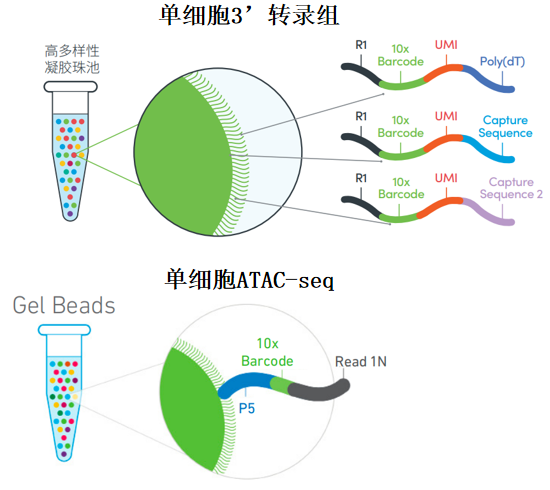
图:凝胶微珠信息
常规单细胞多组学研究往往分步进行:先在同一样本上分别采集不同组学数据,再通过生信方法将它们整合。那么,是否有技术能在同一细胞内同步捕获染色质可及性和基因表达?答案是肯定的!
01. 10xGenomics单细胞多组学
10xGenomics单细胞多组学(scRNA-seq+scATAC-seq)产品的微珠表面有两种捕获序列:一条捕获mRNA,一条捕获染色质可及区段。
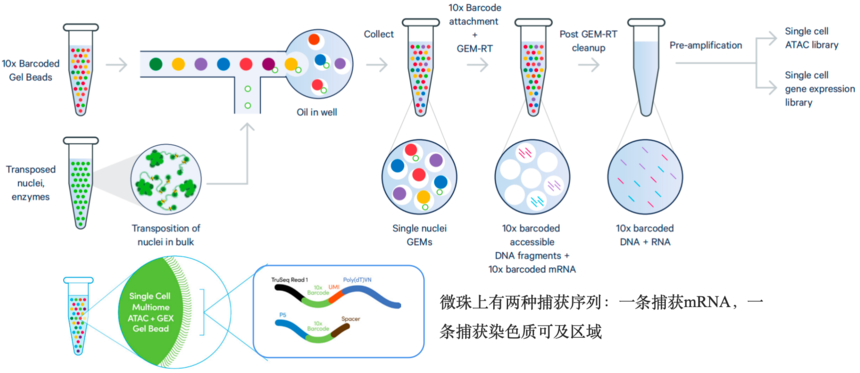
图:10xGenomics单细胞多组学(scRNA-seq+scATAC-seq)
02. 寻因单细胞多组学
国产平台寻因单细胞多组学(scRNA-seq+scATAC-seq)和10xGenomics在主要流程相同,但是微珠表面的序列有所差异。微珠表面只有一种序列,可同时捕获RNA和染色质可及区段。
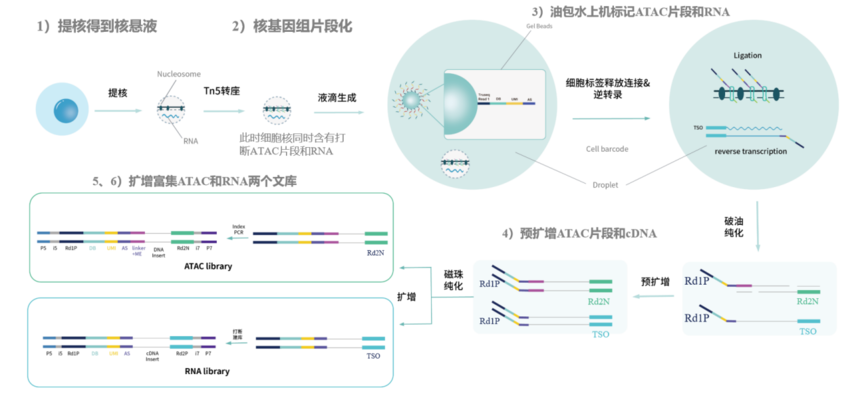
图:寻因单细胞多组学(SeekOne® DD的scRNA-seq+scATAC-seq)
除了以上两个基于微流控平台的单细胞多组学外,还有一个产品可以检测同一个细胞的全基因组和转录组,也值得推荐一波~
03. BioSkryb单细胞多组学ResolveOME
鉴于单细胞中RNA和DNA含量极其有限,实现高效扩增与高质量文库构建至关重要。ResolveOME 技术能够在同一细胞内同步获得高质量的全基因组和转录组多组学数据,为深入解析细胞异质性和分子机制提供了坚实基础。
该技术的核心点在于聚合酶(Isothermal polymerase)和终止子(Termination Base)。经过改造后的DNA聚合酶合成DNA链,掺入一定比例的终止碱基,缩短合成DNA链的长度(长度大致在250-1500bp)。而改造过DNA聚合酶对于长链DNA有偏好性,更容易结合到原始的长链DNA上。这样子它在扩增的过程中始终以原始模版,减少PCR所导致的错误碱基积累,扩增的产物更加均一。
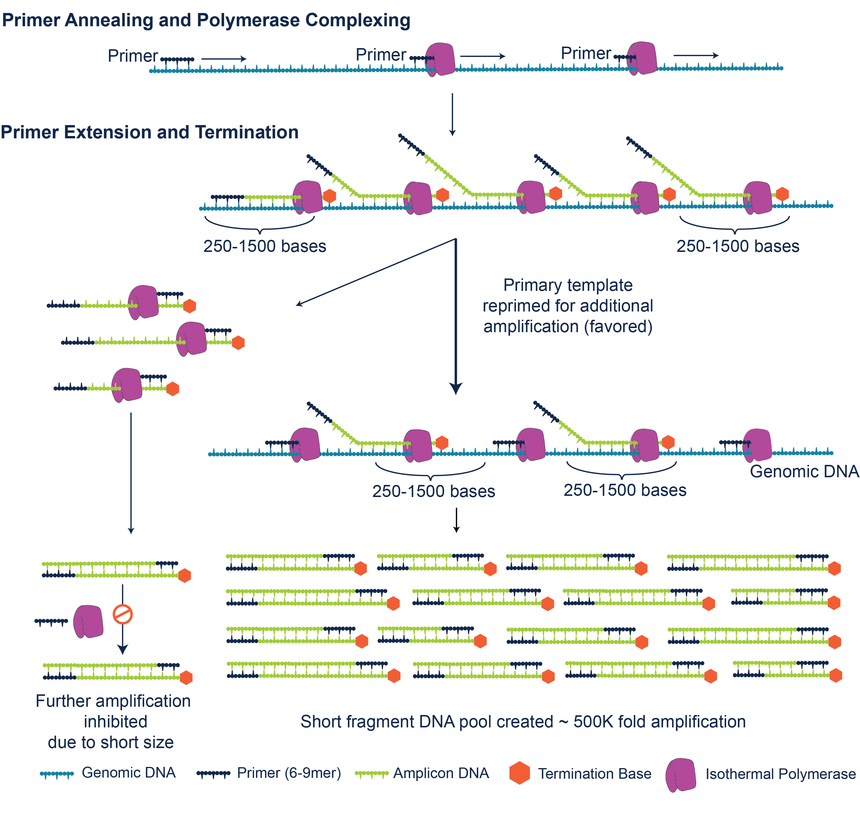
图:技术流程
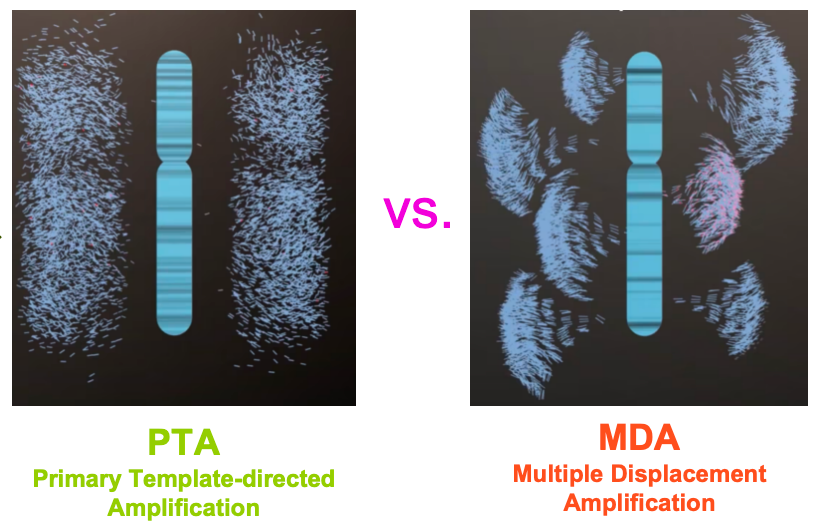
这个扩增方法叫原代模版定向扩增技术(PTA,Primary template-directed amplification)。从下图结果可以看出PTA的扩增更均一。
ResolveOME实验流程是先收集细胞,然后将RNA逆转录为cDNA。进行DNA的扩增后,用有poly(A) 链的磁珠把cDNA与全基因组的DNA进行分离,分开制备文库,再上机测序,这样就可以同时检测DNA和RNA了。
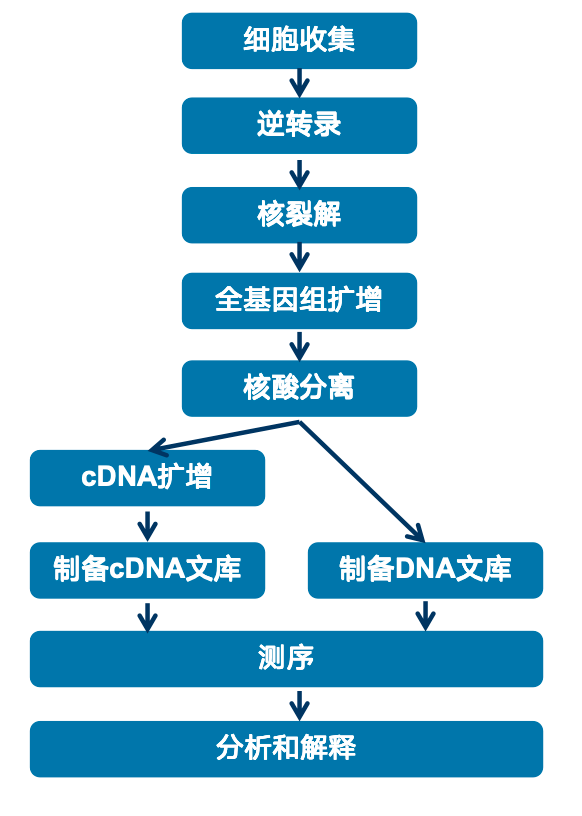
图:ResolveOME技术流程
10xGenomics和寻因的单细胞多组学主要侧重于区分组织异质性的细胞群体多态性分析,而BioSkryb单细胞多组学ResolveOME产品针对微量的单细胞,不需要微流控仪器。
悄悄打个广告,以上单细胞多组学技术爱基均可以提供哦,欢迎有需求的老师咨询哟~
单细胞多组学联合分析
随着单细胞测序技术的不断发展,多组学整合已成为揭示细胞异质性与分子调控网络的利器。通过同时获取基因组、转录组、表观组等多层次数据,单细胞多组学不仅能实现对细胞类型和状态的更加精准、全面注释,还可借助各组学结果的交叉印证提升分析可靠性。
更重要的是,它能够将调控元件链接到靶基因及其上游转录因子,重建基因调控网络,推断细胞发育与分化的动态轨迹;此外,单细胞多组学能敏锐捕捉稀有或瞬态的细胞类型/状态,还可以利用一种组学数据去预测另一种,为深度解析生物过程与疾病机制提供了前所未有的视角和可能性。
01. 细胞注释更精准和全面
早在2019年,一篇发表在Cell上的文章就提出来一个策略——创新性的"锚点"(anchor)策略,用于整合不同模态的单细胞数据。基于Seurat,通过典型相关分析和锚点识别来整合不同单细胞实验的数据集,以改善数据分析的准确性。
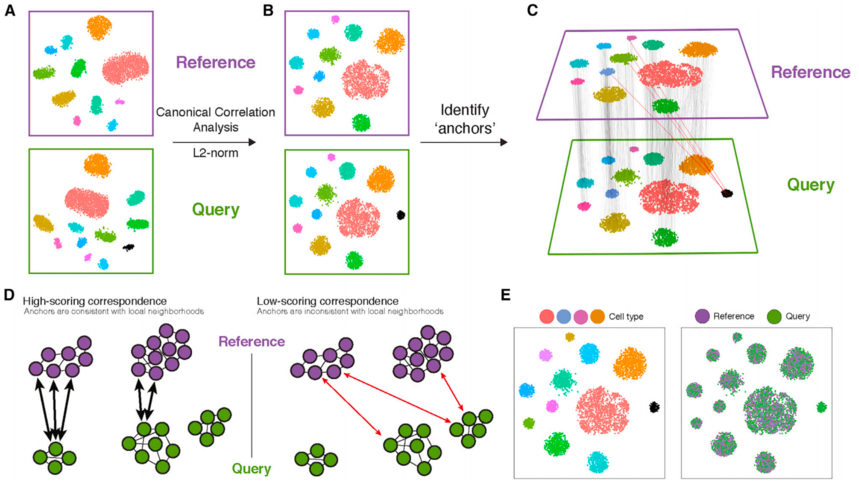
图:Seurat v3 中参考“组装”集成的示意概述
02. 多组学结果互相印证
a. 基因表达数据和开放性数据印证(scRNA-seq+scATAC-seq)
scRNA-seq+scATAC-seq联合分析的主要逻辑有:开放染色质区域与基因表达具相关性;远端和近端染色质开放区域都有可能调控下游的基因表达;基因组开放性变化的区域可能与哪些转录因子结合以调控基因表达。
在单细胞转录组和单细胞ATAC-seq分析结果中,经常可以看到两个组学的相关性分析(下图)。单细胞转录组侧重于测量转录本的表达水平,而单细胞ATAC-seq则能帮助我们了解染色质可及性甚至潜在的调控元件活性。将二者做关联分析(如将同一个细胞群或同一细胞类型在RNA与ATAC水平做相关),可以更全面地理解基因调控网络、转录因子结合位点以及这些因素对基因表达的最终贡献。这样既能相互印证某些结果的可靠性,又能从不同维度揭示转录调控机制,在多组学整合分析中尤为常见。
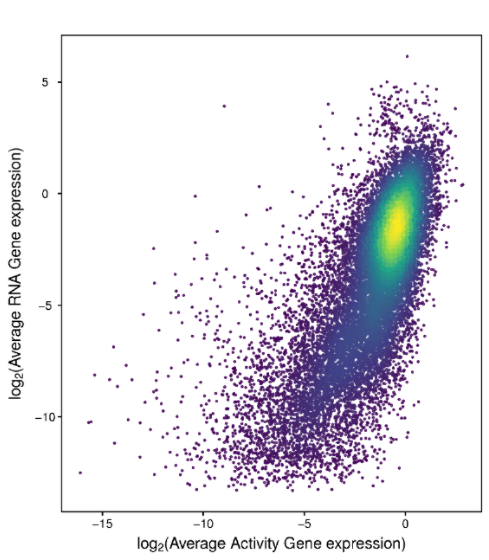
图:scRNA-seq和scATAC的相关性
在scATAC-seq数据中,对于每一个细胞,通过基因及基因上游2kb内的peak丰度量化每个基因在每个细胞中的基因开放性;即在这个区域内,peak丰度越高,基因就越有可能受到转录因子调控或与RNA聚合酶结合,基因开放性越高。对于每一个样本,我们计算平均基因开放性(基于scATAC-seq)和平均基因表达量(基于scRNA-seq),并进行相关性分析,使用pearson相关性系数来衡量每个样本两组学的相关性强弱。
b. 基因表达数据和组蛋白修饰数据印证(scRNA-seq+scCUT&Tag)
scRNA-seq+scCUT&Tag联合分析的主要逻辑有:组蛋白修饰与基因表达呈正/负相关;组蛋白修饰(比如组蛋白乙酰化类)使得染色质结构松散,促进基因表达。
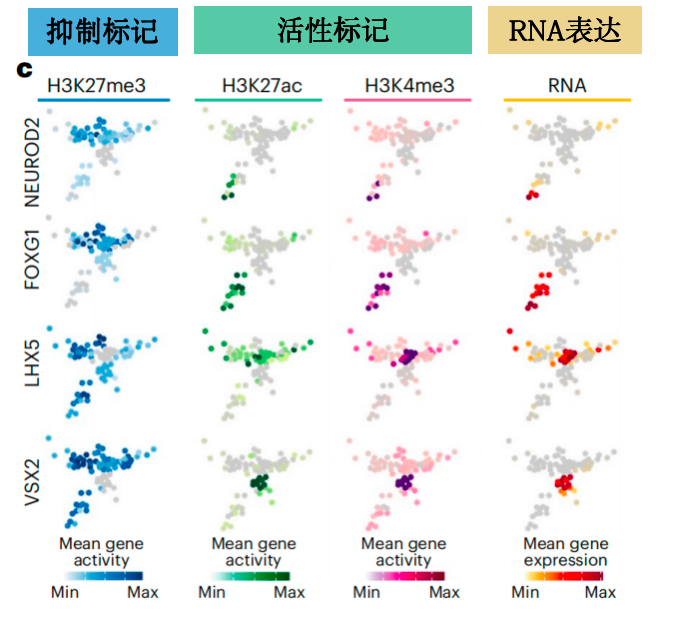
图:scRNA-seq+scCUT&Tag:区域特异性转录因子的RNA表达和基因活性(基因及其扩展的启动子区域上游+2 kb的组蛋白修饰丰度)互相印证[3]。
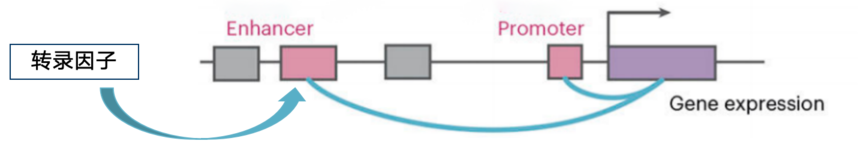
03. 调控元件链接到靶基因和上游转录因子
a. 链接靶基因
SCENT(单细胞增强子靶基因图谱)可以用于建模单细胞或细胞核多模态RNA测序和ATAC测序数据中增强子染色质开放性与基因表达之间的关联。该方法通过统计方法(如泊松回归)来测试每个峰的可及性是否与目标基因的表达水平显著相关。如果某个峰的可及性与基因表达显著相关,那么这个峰可能是一个功能性的调控元件,能够影响该基因的表达。
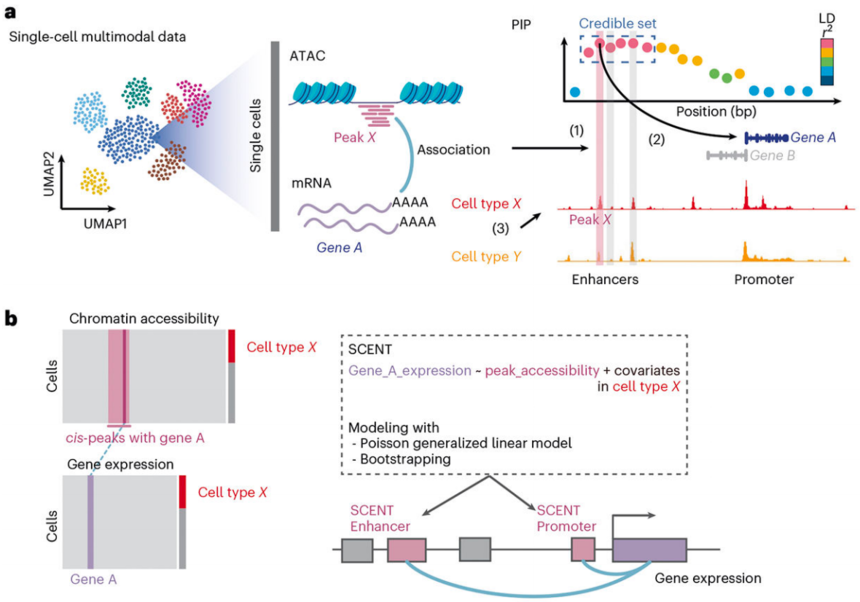
图:SCENT模型(单细胞增强子靶基因图谱):单细胞多组学ATAC + RNA 多模态数据的模型[2]
b. 链接转录因子
单细胞多组学还可以将可能与增强子互相作用以激活靶基因的转录因子结合起来,可以用一些工具比如Scenic+、REUNION等。逻辑:scRNA-seq提供靶基因表达数据和转录因子活性数据,scATAC-seq提供增强子信息;然后将这些数据相关联起来。调控元件链接靶基因和转录因子后,就可以基于这些分析构建相关的调控网络。
04. 解析发育轨迹
单细胞表观多组学可助力谱系预激研究。在发育过程中基因“被打开”之前,调控染色质的可及性会增加。类似地,组蛋白修饰标记在谱系预激中起到“开门锁”的作用。这些修饰能改变组蛋白对DNA的结合方式,从而影响染色质的松紧程度和周围调控元件的可及性。
scRNA-seq和scATAC-seq联合分析以下图为例,从过度扩充细胞到角质/皮层细胞的分化拟时间,追踪了Wnt3基因位点附近各个峰的可及性变化。研究发现Wnt3 染色质调控区域中的峰值依次激活,增强子峰值的激活比启动子早得多,随后是新生RNA表达(由内含子counts估算)的激活,最后是成熟RNA表达(由外显子counts估算)。
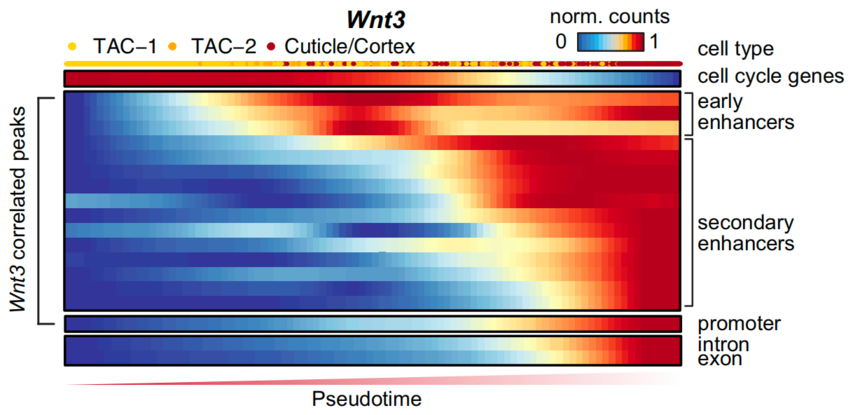
图:增强子中的谱系预激先于染色质调控区域中的基因表达[4]
在一项神经发育研究中,研究团队以大脑类器官为模型,联合开展了 scCUT&Tag(检测组蛋白修饰)、scATAC-seq(检测染色质可及性)与 scRNA-seq(检测转录组)三种单细胞多组学实验。从可视化结果上我们可以看到激活组蛋白修饰H3K27ac和H3K4me3在检测到染色质可及性增加之前不久就已经建立,随后是RNA表达。这意味着神经发生过程中,表观遗传激活先于基因表达。
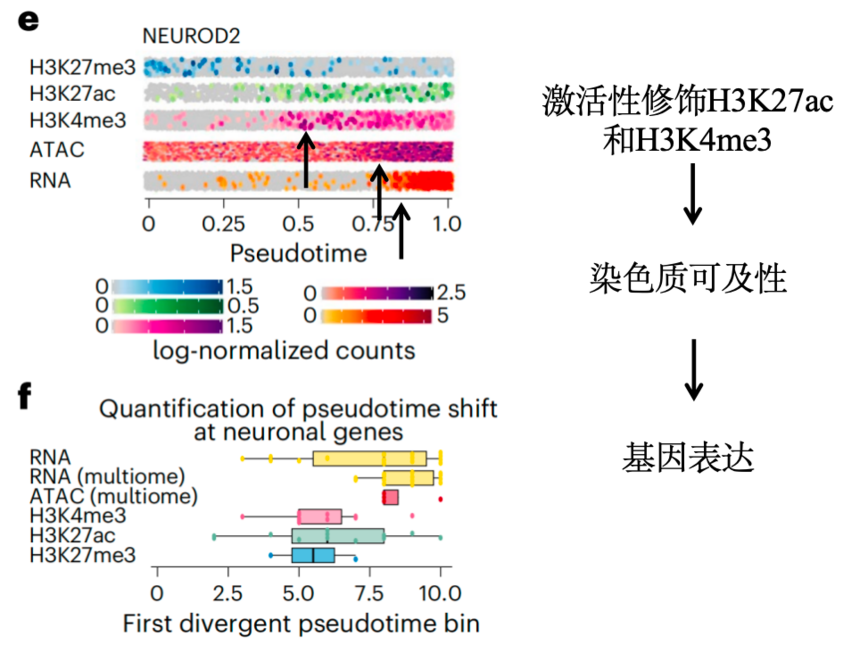
图:量化神经元基因的拟时间[5]
05. 发现稀有/瞬态的细胞类型/状态
部分细胞群体在整体细胞池中占比较小,或者存在短暂转折状态,单一模态往往难以捕捉到这些罕见或瞬态事件。通过多组学分析,多个层面的数据可以共同指向这些细胞群的特殊性,例如即使RNA表达变化微弱,但其染色质状态或特定的蛋白表达可能尤其明显。这种综合证据能够帮助我们识别并验证这些重要而难找的细胞类型或状态,为疾病发生机制研究和精准治疗提供新线索。
MarsGT是一个专为识别单细胞多组学数据中罕见细胞群体设计的工具。它采用基于概率的异构图转换器方法,能够同时鉴定主要和罕见细胞群体,并建立其基因调控网络。
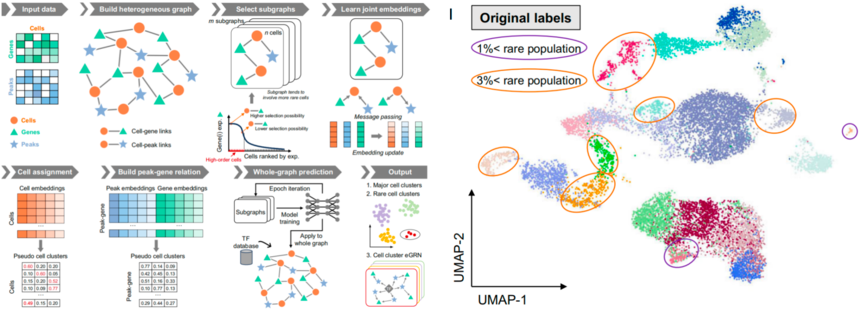
图:MarsGT流程[6]
06. 用一个模态预测另外一个模态
SCARlink (Single-Cell ATAC + RNA linking) 是一种创新的单细胞多组学分析工具,专为整合单细胞ATAC-seq和RNA-seq数据而设计,通过多组学测序方法预测基因表达并将增强子连接到目标基因。SCARlink采用正则化泊松回归模型,将单细胞中的染色质可及性作为输入来预测基因表达,它还可以用于识别候选增强子。
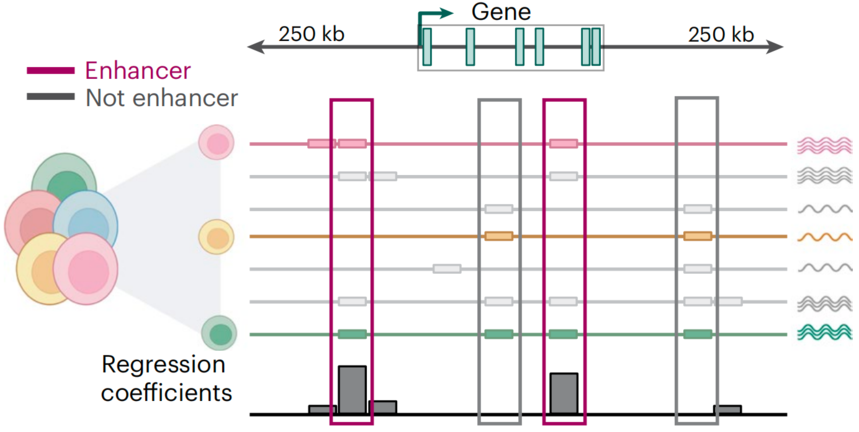
图:SCARlink准确地预测了染色质可及性中的单细胞基因表达[7]。
以单细胞ATAC测序在基因位点的计数数据作为输入(超500bp tiles, 覆盖基因体上游 250 kb 至下游 250 kb 的区域),采用正则化泊松回归方法预测基因的单细胞表达水平。
MarsGT和SCARlink这两个工具均是2024年发表的,在单细胞多组学里面是较为新兴的联合分析方法。随着相关联合分析工具的开发,相信还会有更多的应用可能性。
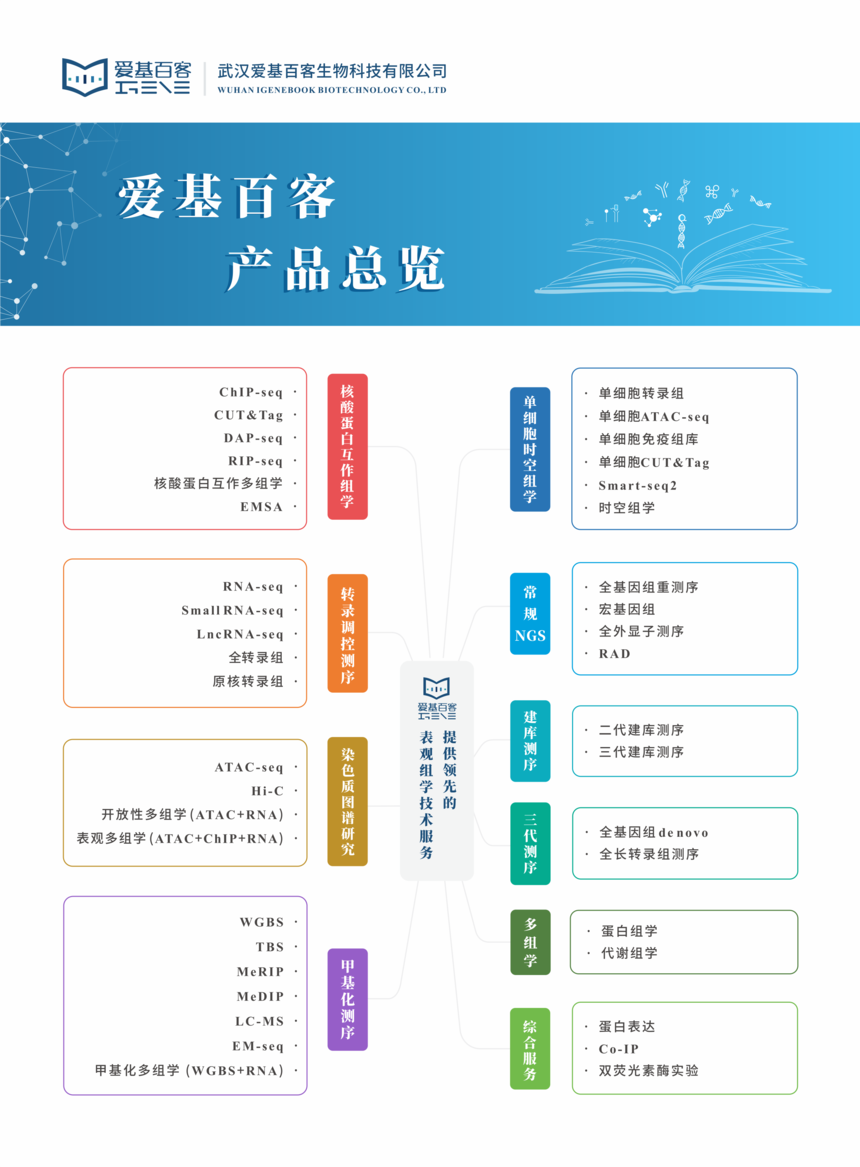
爱基百客致力于提供领先的单细胞测序技术服务。如您有单细胞技术服务需求,欢迎联系我们~
-
参考文献
【1】Laisné M, Lupien M, Vallot C. Epigenomic heterogeneity as a source of tumour evolution[J]. Nature Reviews Cancer, 2025, 25(1): 7-26.
【2】Sakaue S, Weinand K, Isaac S, et al. Tissue-specific enhancer–gene maps from multimodal single-cell data identify causal disease alleles[J]. Nature genetics, 2024, 56(4): 615-626.
【3】Zenk F, Fleck J S, Jansen S M J, et al. Single-cell epigenomic reconstruction of developmental trajectories from pluripotency in human neural organoid systems[J]. Nature Neuroscience, 2024, 27(7): 1376-1386.
【4】Ma S, Zhang B, LaFave L M, et al. Chromatin potential identified by shared single-cell profiling of RNA and chromatin[J]. Cell, 2020, 183(4): 1103-1116. e20.
【5】Zenk F, Fleck J S, Jansen S M J, et al. Single-cell epigenomic reconstruction of developmental trajectories from pluripotency in human neural organoid systems[J]. Nature Neuroscience, 2024, 27(7): 1376-1386.
【6】Wang X, Duan M, Li J, et al. MarsGT: Multi-omics analysis for rare population inference using single-cell graph transformer[J]. Nature Communications, 2024, 15(1): 338.
【7】Mitra S, Malik R, Wong W, et al. Single-cell multi-ome regression models identify functional and disease-associated enhancers and enable chromatin potential analysis[J]. Nature genetics, 2024, 56(4): 627-636.


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









