






 本文通过结合单细胞测序、空间转录组和染色质可及性技术,揭示了心肌梗死后心脏重塑过程中基因调控网络的变化,特别是RUNX1在肌成纤维细胞分化中的关键作用。研究提供了全面的心肌梗死分子图谱,推动心脏病机制和治疗研究的发展。
本文通过结合单细胞测序、空间转录组和染色质可及性技术,揭示了心肌梗死后心脏重塑过程中基因调控网络的变化,特别是RUNX1在肌成纤维细胞分化中的关键作用。研究提供了全面的心肌梗死分子图谱,推动心脏病机制和治疗研究的发展。
英文题目:Spatial multi-omic map of human myocardial infarction
发表时间:2020年12月
单细胞测序技术分辨率和检测通量的大幅度提高,使得研究者能够在单细胞分辨率上得到细胞之间的异质性,而空间转录组则为研究者提供了组织中细胞所处的空间信息,及组织中不同区域的细胞构成和基因表达状态,但由于最新的空间转录组技术还达不到单细胞的分辨率,因此目前比较常见的是空间转录组联合单细胞测序技术一起应用。 冠状动脉心脏病导致的急性心肌梗死(MI)是心血管疾病死亡的最大原因,也是全球所有死亡的主要原因。MI的急性治疗取得了巨大进展,主要集中在经皮冠状动脉介入治疗,从而降低了急性死亡率。然而,心肌梗死后左心室重塑引起的发病率和死亡率仍然高得不能接受。心肌梗死后的心脏重塑包括免疫细胞招募和梗死区域的划分,随后是组织消化、吞噬、肌成纤维细胞活化、疤痕形成和新生血管形成。理解从急性缺血事件到慢性心脏瘢痕形成的心脏重构过程的确切细胞和分子机制将是开发新的治疗方法的关键。 在这项研究中,作者使用了单细胞基因表达和染色质可及性技术的结合,以及空间分辨转录组学来研究基因调控的细胞类型特异性变化,提供了心肌梗死后心脏重构的整合图。这揭示了在健康和疾病中控制特定心脏细胞类型的基因调控网络。作者将这些信息投射到特定的组织位置,从而使作者能够在空间上绘制控制基因调控的假定增强子,例如心肌边界区。这反过来又使作者能够获得新的见解,了解在心脏瘢痕形成过程中驱动成纤维细胞向肌成纤维细胞分化的基因调控程序。实验结果提供了一个全面的空间解决表征基因调控的人心脏在稳态和心肌梗死后,并****鉴定和验证了驱动心脏纤维化的成纤维细胞向肌成纤维细胞分化的机制。本文的研究提供了一个完整的人心肌梗死的分子图谱,并为心脏疾病的机制和治疗研究的进展提供了参考。

研究结论
本文研究提供了关于心脏细胞类型及其对人类心脏缺血损伤的反应的详细见解。作者观察到在局部细胞因子表达的指导下,局部免疫细胞群的流入可以早期划分梗死区域。在受伤心肌周围的边界区,受伤和未受伤的细胞类型之间确实有明显的界限。不同位置的心肌细胞边界显示出不同的基因表达模式和基因调控谱,包括增强子可及性。心肌梗死后的晚期重塑是由成纤维细胞纤维化驱动的,在明显的疤痕中,疤痕周围有新血管生成的区域。本实验的数据为MI后人类心脏肌成纤维细胞分化提供了新的见解,独特的基因表达和基因调控程序驱动这一过程,包括RUNX1作为TGFβ信号的放大器。提供了一种新的策略将单细胞技术和空间转录组技术结合开展时空多维解决生物问题,为肿瘤疾病的预防治疗和机制探讨提供了技术平台。
实验材料
材料:4例心肌梗死患者、1例健康供者,8例左心室组织标本;非移植供体心脏(对照,C)与急性心肌梗死患者的坏死中心(缺血区,IZ)、边界区(BZ)和未受影响的左心室心肌(远端区,RZ)作为对照,2例心肌梗死(心肌纤维化区FZ)后晚期(3个月和12年)的人心脏;获得每个心脏标本10 m的冷冻切片,并从紧邻冷冻切片的其余标本中分离细胞核,随后使用荧光活化核分选(FANS)进行snRNA-和snATAC-seq
方法:10x 单细胞核转录组测序;单细胞ATAC-seq;10x visium 空间转录组等
文章亮点
1. ScRNA-seq揭示了人类心脏细胞异质性;
2. 空间转录组探讨心肌梗死过程揭示心肌重构过程;3. 染色质开放性和基因表达调控关系;4. snRNA-seq+scATAC-seq+空间转录组联合锁定心脏肌纤维细胞分化的调节因子RUNX1
![]()
研究结果
1. 单细胞转录组和染色质景观揭示了人类心脏细胞类型的异质性研究人员使用单细胞核转录组技术(snRNA-seq)通过降维聚类识别了24个簇细胞群,构建一个整体的肿瘤生态位图谱。
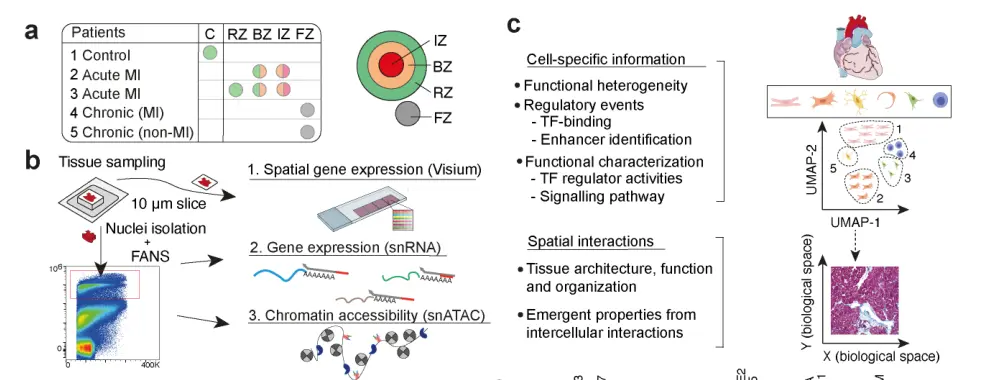
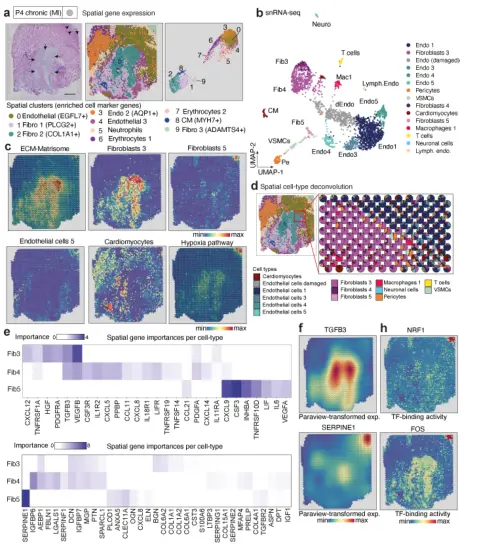
图1. 单细胞核+单细胞ATAC+空间转录组多组学图谱snRNA-seq共获得40530个核转录组,平均每个核1335个基因;scATAC-seq共检测到18213个核的开放染色质,平均每个核24333个片段(n=7);空间转录组平均每个样本包含3874.5个spot,每个spot包含1813个基因。进一步对scRNA-Seq进行细胞类型注释共鉴定到10种主要的细胞类型和几个亚群(总共n=24个簇),而scATAC-seq能够鉴定到在snRNA-seq数据中确定的10种主要细胞类型中的8种细胞类群。空间样本的差异分析及富集分析能清晰地反映心肌梗死后生物过程的已知区域。虽然大多数标本中存在主要的细胞类型,但不同的细胞类型仅在单个标本中被识别,可能反映了损伤或修复过程中不同的细胞状态。 在急性心肌梗死缺血区,观察到与先天免疫系统、中性粒细胞脱颗粒和程序性细胞死亡以及纤维化和肌肉收缩过程的消耗相关的空间可变基因表达的富集(图1h)。同样,慢性重塑心脏(心肌梗死后的晚期)显示了与细胞外基质(ECM)蛋白多糖、糖蛋白和其他基质体成分相关的空间可变基因的富集,与这些标本中捕获的预期纤维化过程一致。边界区标本显示了丰富的与线粒体复合物I生物发生和丙酮酸代谢/柠檬酸TCA循环相关的基因,这两者都证实了该区域对损伤的反应和潜在的氧化还原状态和代谢的改变。在对照组和远端区标本中,与肌肉收缩相关的空间可变基因的富集,这与这些样本中健康心肌细胞的过度表达有关。总的来说,这一分析证实了空间数据清楚地反映了心肌梗死后已知的生物过程区域。
2. 健康人心脏的多组学综合分析
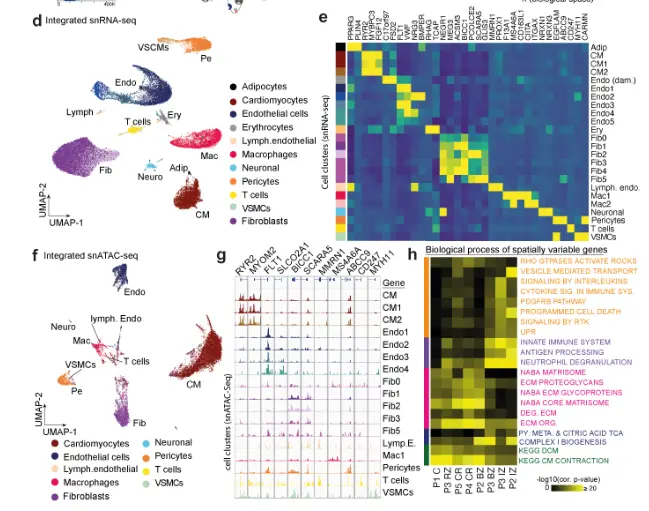
首先将对照人类心脏样本的整合数据作为参考数据集进行深度表征,从snRNA-seq (n = 8,335)和snATAC-seq (n = 3,849)中分别鉴定出12种细胞类型(图2a-b)。通过对snATAC-seq数据的转录因子足迹分析,揭示了已知细胞特异性转录因子(TF)的结合活性,如心肌细胞中的MEF2C、巨噬细胞中的ETV6和内皮细胞中的SOX8(图2c)。这些TFs的活性通过靶基因的细胞型特异性表达得到进一步证实(图2d)。将同一左心室心脏组织的空间转录组数据聚类,得到11个分子差异的聚类(图2e)。差异基因表达分析显示心肌细胞是个体空间集群的主要转录组贡献者(图2e)。值得注意的是,标记基因允许在不同区域识别常见的细胞类型,如心肌细胞,但也可以识别罕见的心肌细胞群,如血管平滑肌细胞(在snRNA-seq数据中占1%的细胞)。有趣的是,肥大细胞(CPA3+)在一个独特的空间聚类中被识别出来(聚类9,图2e),而它们在snRNA-或snATAC-seq数据集中均未被检测到(图2a-b)。在snRNA-seq和空间转录组数据中比较顶部差异表达的基因证实了空间注释图。
接下来,建立了两个成纤维细胞亚簇成纤维细胞1+2的空间分辨表达模型(图2h),并鉴定了几种可以解释成纤维细胞2位置的细胞因子。有趣的是,该模型显示,与巨噬细胞亚型2相关的IL13RA1的局部表达预测成纤维细胞2标记物FBN1的表达,提示在心脏稳态中,成纤维细胞和巨噬细胞的特定亚群之间可能存在信号传导(图2h,)。
![]()
3. 不同的基因表达调控来区分缺血区
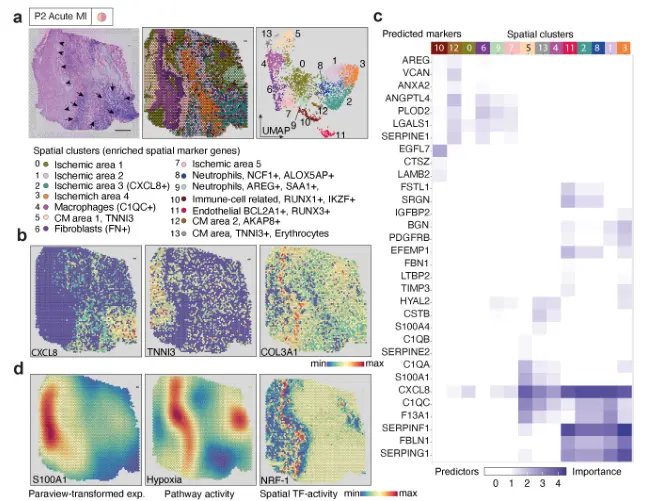
研究者在急性人类心肌梗死组织缺血区标本的snRNA-seq和snATAC-seq数据中鉴定到4种不同的细胞类群。研究者在空间标本中心确定了一个核心缺血区(cluster 3),对该区域及周边的基因表达分析发现其表达特征与病理分区一致。
为了探索与空间区域相关的TF,利用细胞类型评分将TF结合活性从snATAC- seq数据映射到空间。观察到局部增加的NRF-1结合活性在该区域也显示了高缺氧信号(图3d)和胶原表达(图3b)。进一步证实了空间TF结合活性与特定NRF- 1靶基因的表达相对应。NRF-1调节编码呼吸链亚基的基因和参与线粒体功能的其他基因的表达,与细胞对缺氧的反应一致
第二个急性心肌梗死标本缺血区的空间转录组数据显示出相似的界限。与同一心脏标本的偏远区域相比,这些区域的预测特征保持不变。在缺血区观察到新血管生成的早期迹象(簇2),EGFL7和SOX4表达,以及一个空间心肌细胞簇显示内质网应激标记基因表达24,如HERPUD1(簇4)。总之,snATAC-seq与空间转录组联合分析数据表明,在缺血相关细胞死亡的响应中有不同的空间基因调控,基因调控驱动急性心脏损伤反应。
4. 边缘区心肌细胞亚群的空间分布特征心肌梗死的边缘区特别值得关注,因为该区域的空间重构与心功能的恢复密不可分,研究者在样本中心确定了一个特定的空间过渡区(聚类1)。该区域将样本左上角和右下角的不同基因表达区分开(图4a)。从snRNA-seq (n = 6,081)中鉴定出13种细胞类型,从snATAC-seq (n = 3,101)中鉴定出8种细胞类型(图4b-c),两个数据集都识别了两个不同的心肌细胞群(心肌细胞1和心肌细胞2)(图4b-c)。心肌细胞1仅位于过渡区(损伤区)下方,而心肌细胞2主要位于过渡区上方(图4d)。scRNA-seq数据的差异基因表达分析显示心肌细胞中NPPB、ANKRD1和MYO18B显著上调1(图4d)。据报道,MI后NPPB和ANKRD1均在边界区上调。空间基因表达数据的通路分析表明,损伤区(右下)TGFβ活性增加,而缺氧活性的均匀分布。总之,分析表明,心肌细胞亚群在损伤、炎症和重塑的不同区域的边界区有空间上不同的基因表达和调控。
5. 心肌梗死后的重构
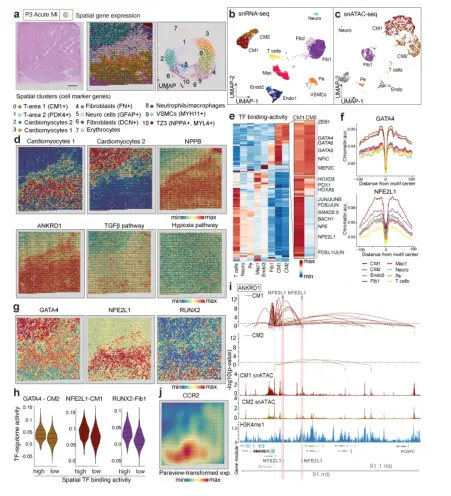
心肌梗死后瘢痕形成对心脏组织完整性很重要,因为瘢痕形成失败会导致心室破裂和死亡。空间转录组数据的聚类显示,这两个疤痕具有明显的聚类(聚类2和聚类9),并被反映新血管生成的更大面积的内皮细胞隔开(图5a)。从snRNA-seq (n = 2,869)和scATAC-seq (n = 1,605)中鉴定出不同的成纤维细胞和内皮细胞亚群,这些亚群在两个瘢痕上显示出有趣的空间分布(图5b-c)。
空间特征的差异分析显示,JAK-STAT和TGFβ活性增加,这两种都是纤维化重塑的重要途径。在成纤维细胞3富集的区域(空间簇2)。富含成纤维细胞5的区域也包含损伤的内皮细胞和增加的缺氧途径活性(图5d)。作者观察到TGFβ3、PDGFRa和PDGFA的局部表达是成纤维细胞3存在的高度重要的预测因子,成纤维细胞3在瘢痕中心区域丰富(图5e-f)。SERPINE1 (PAI-1)与组织中的活性瘢痕相关33,在簇9中显示出高度重要性,并与较高的缺氧和VEGF-A信号有关(图5e-f)。相比之下,中央瘢痕区(空间簇9)也与更高的NRF-1和TF - FOS结合活性相关(图5h)。作者使用平行的snATAC- seq数据进一步验证了这一点,该数据显示TGFβ通路下游SMAD2/3具有更高的结合活性(扩展数据图12h)。总之,这些数据表明了两个瘢痕区域之间的时间差异,其中一个是较年轻的瘢痕(右上),具有强缺氧信号和明显的成纤维细胞亚群,另一个是较老的慢性大瘢痕,涉及FOS, SMAD2/3,NRF1和RUNX1的高基质产生。
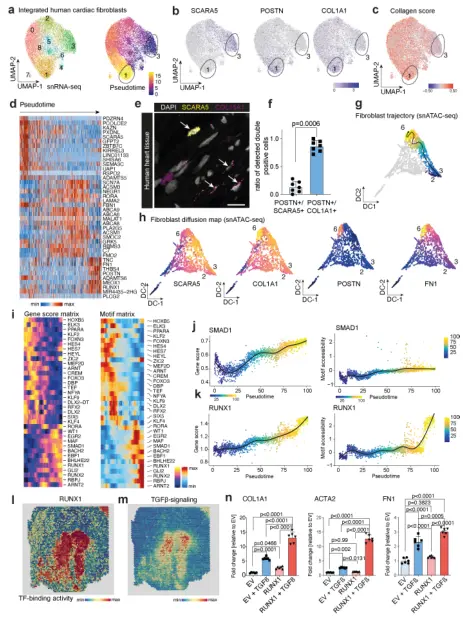
6. 轨迹分析显示RUNX1是人心脏肌成纤维细胞分化的调节因子
为了解剖肌成纤维细胞分化的机制,从整合的snRNA-seq数据集中重新聚集了所有成纤维细胞,并鉴定出9个亚簇(图6a),表明心脏间质存在未被重视的异质性。拟时序分析显示,完整的成纤维细胞分化梯度起源于簇3,终止于簇1(图6a,)。发现SCARA5作为人类肾脏成纤维细胞的标记物,拟时序分析推测SCARA5+细胞向POSTN+细胞分化,对拟时序轨迹基因、通路和Go-Term(基因本体)进行了分类,证明了整合素信号和ECM通路的晚期富集与成纤维细胞到肌成纤维细胞的分化是一致的。并证明SCARA5 +成纤维细胞是肾脏肌成纤维细胞的一个来源。因此,在拟时序分析中使用SCARA5表达量最高的细胞(簇3)作为根细胞。骨膜素(POSTN)是肌成纤维细胞分化的标志,在人和小鼠之间似乎是保守的,并在肾脏纤维化中发挥作用。数据表明RUNX1是肌成纤维细胞分化的重要驱动因子,并且通过放大TGFβ信号发挥作用。结合已知的RUNX1在心肌细胞中的作用,该转铁蛋白可能成为治疗心肌梗死后心脏重塑的一个非常有前景的靶点。
总结
在多细胞器官中,如人类心脏,细胞的功能依赖于相邻的单个细胞类型之间相互作用的平衡,这导致了组织稳态。单细胞技术可以描绘不同细胞类型的分子异质性及其在疾病期间的变化。然而,如果没有空间背景,就不清楚这些不同的细胞类型如何协调组织功能。在这里,整合了空间转录组学、单细胞基因表达和染色质可及性数据,提供了心肌梗死早期和晚期与对照心脏(非移植供体心脏)相比的全面资源图。利用空间转录组学数据,可以研究细胞系和功能之间的联系,这是单细胞技术无法实现的。利用空间住转录组和单细胞联合的策略提高细胞类型组成的空间分辨率,并通过识别损伤、修复和重塑的不同细胞区域,提供对心脏转录组和表观基因组的空间分辨率洞察力。鉴定并验证了成纤维细胞向肌成纤维细胞分化从而驱动心脏纤维化的机制。提供了一个完整的人类心肌梗死分子图谱,为进一步研究心脏病的机制和治疗提供了参考。
多多学习

















 2236
2236

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









