







Single-cell eQTL models reveal dynamic T cell state dependence of disease loci
留意更多内容,欢迎关注微信公众号:组学之心
解读文章有理解错误的地方劳请指出!
研究团队及研究单位
:::: column
::: column-left
Soumya Raychaudhuri–Harvard Medical School

:::
::: column-right
D. Branch Moody–Harvard Medical School

:::
::::
文章简介
背景
全基因组关联研究(GWAS)已经将非编码变异与调控区联系起来,这些变异可能通过调节基因表达导致疾病。然而,这些变异对基因表达的影响(eQTL)并不能完全解释其致病性,因为eQTL的效应大小会因细胞状态、细胞类型组成和环境变化而不同。
特别是在T细胞中,其功能状态由表面标志物、细胞因子、转录因子或转录组程序定义,这些状态是连续且动态的。例如,TH17细胞可以转变为与结核病相关的TH17/1细胞。在单细胞水平上,这些状态可以被捕捉,但许多单细胞eQTL研究通过将离散细胞群体聚合,并使用pseudo-bulk线性模型来分析状态依赖性,这种分析方法限制了对连续转录景观的精确划分。
研究主要内容
研究主要在记忆T细胞中以单细胞分辨率研究了eQTL,使用来自259名秘鲁人的50万个未刺激的记忆T细胞数据,利用低维嵌入来表示多模态单细胞测定的细胞状态异质性。通过分解每个细胞中的多种状态,以单细胞分辨率剖析“细胞状态”关系密切的 eQTL 效应。
- 在6,511个顺式eQTL中,大约三分之一的效应受连续多模态定义的细胞状态影响,例如细胞毒性和调节能力。
- 在某些基因座中,独立的eQTL变体具有相反的细胞状态关系。
- 细胞状态依赖性eQTL中富集了与自身免疫相关的变异,包括 ORMDL3 和 CTLA4 附近的类风湿性关节炎风险变异,表明细胞状态背景对于理解eQTL的致病性至关重要。
- 连续细胞状态比传统的离散分类(例如 CD4+ 与 CD8+)更能解释eQTL的变化,这表明以单细胞分辨率建模eQTL和细胞状态有助于加深对功能异质细胞类型中基因调控的理解。
研究结果
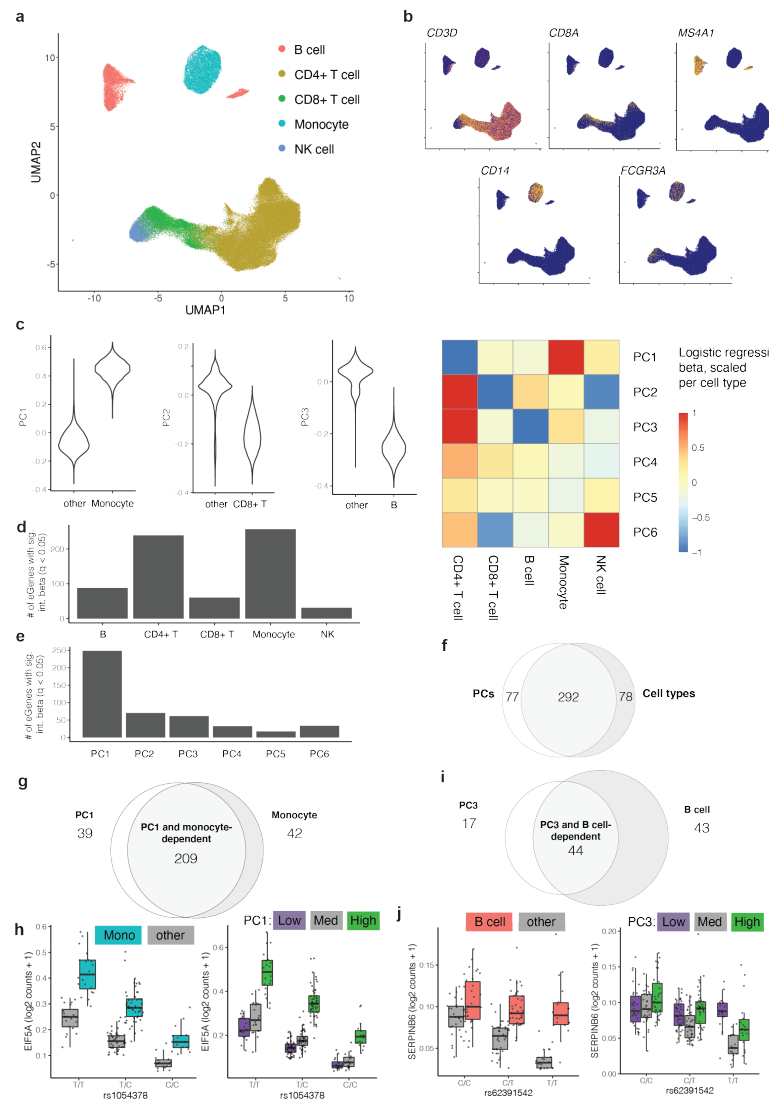
1.秘鲁人样本中记忆 T 细胞的 eQTL
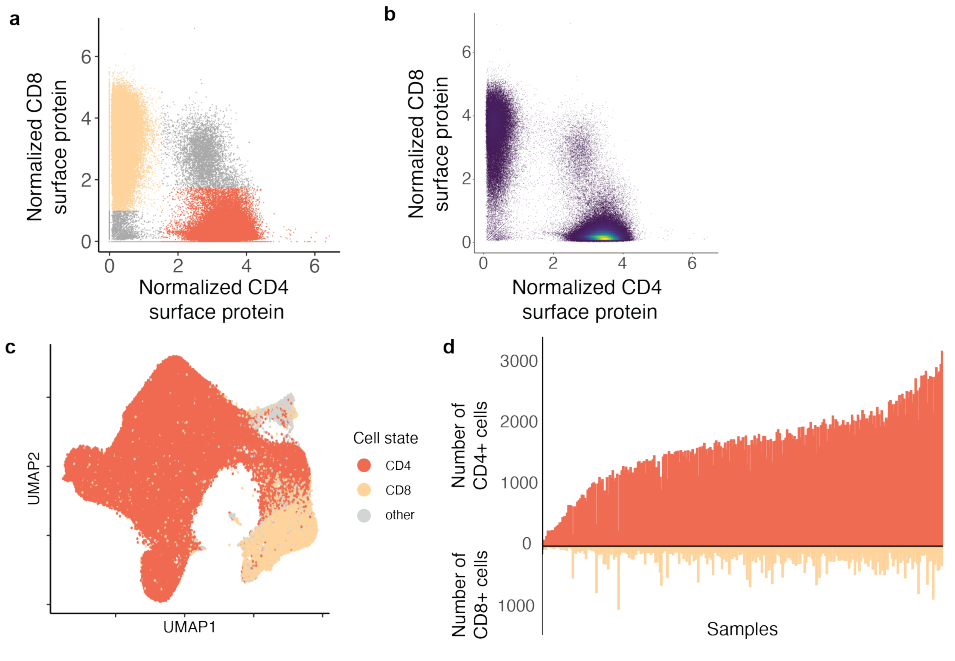
研究使用CITE-seq对 259 名健康秘鲁人分离的 500,089 个记忆 T 细胞进行的转录组和表位细胞索引,这些个体之前曾被结核分枝杆菌感染。数据质量如下图所示:
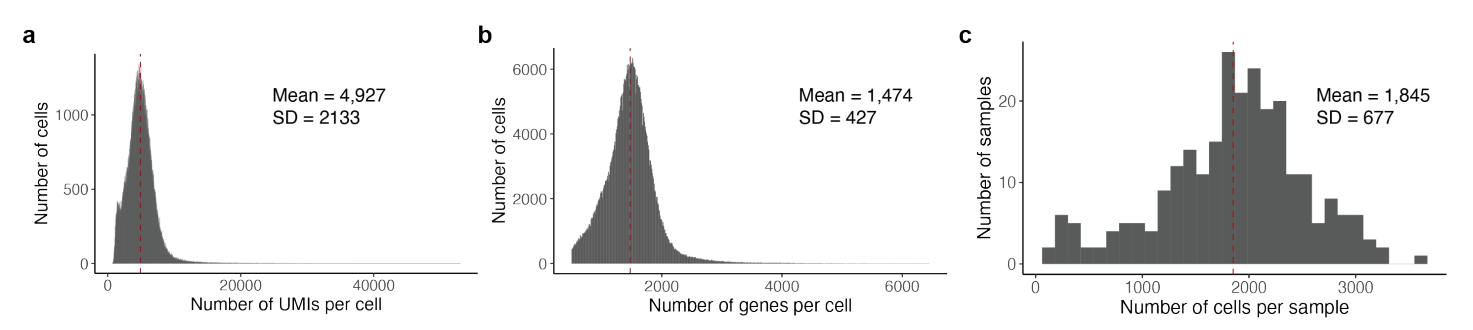
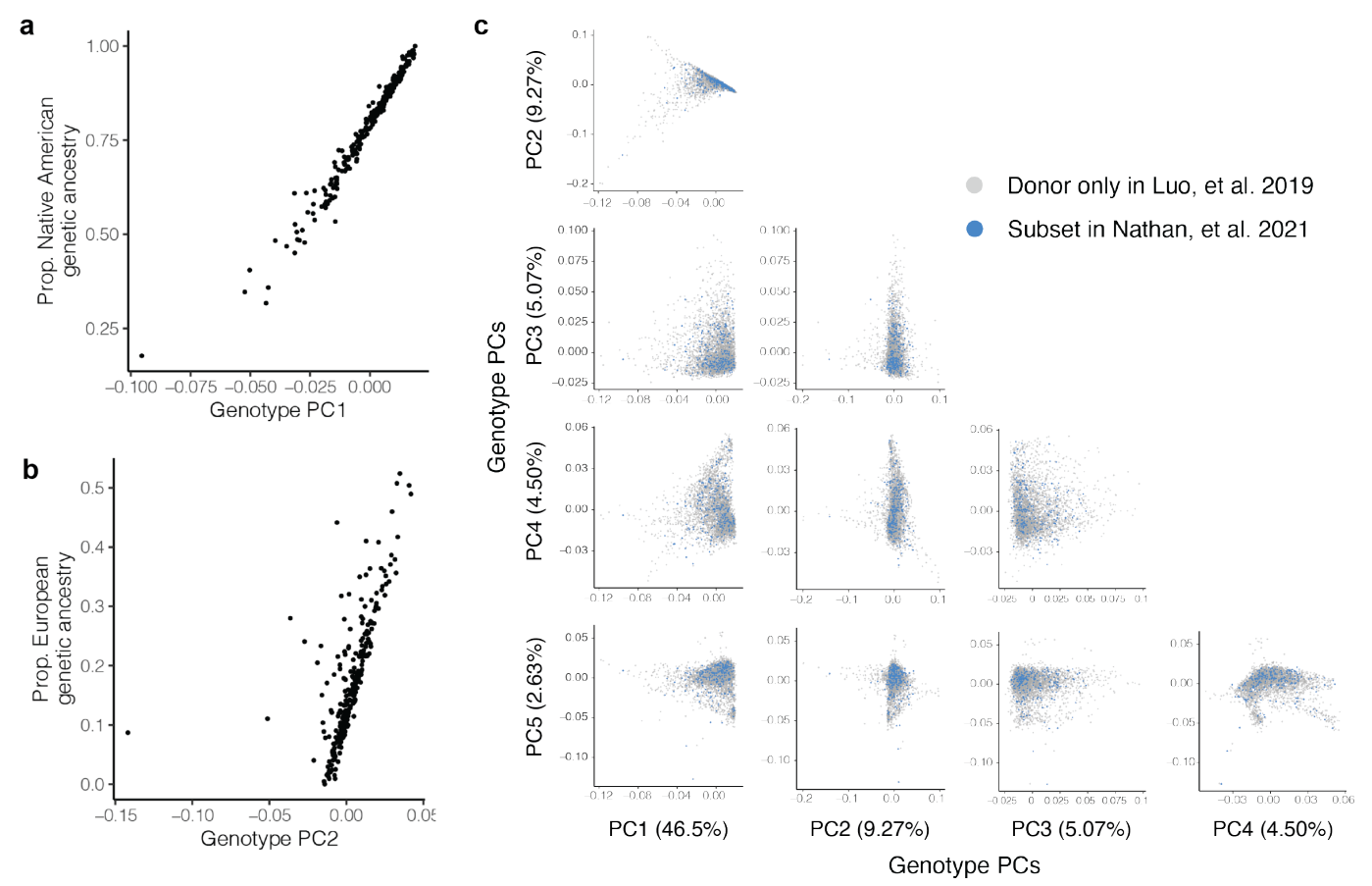
从下图的a和b可见,随着Genotype PC1/PC2的值增加,美洲原住民遗传血统/欧洲遗传血统的比例也在增加,展现了它们之间的强相关性,说明样本是从更大的混合美洲原住民和欧洲遗传血统的基因分型个体群中选出的。经过质量控制,研究分析了 5,486,956 个遗传变异,可见在识别影响基因表达的eQTL时考虑了遗传背景的多样性对结果的影响:
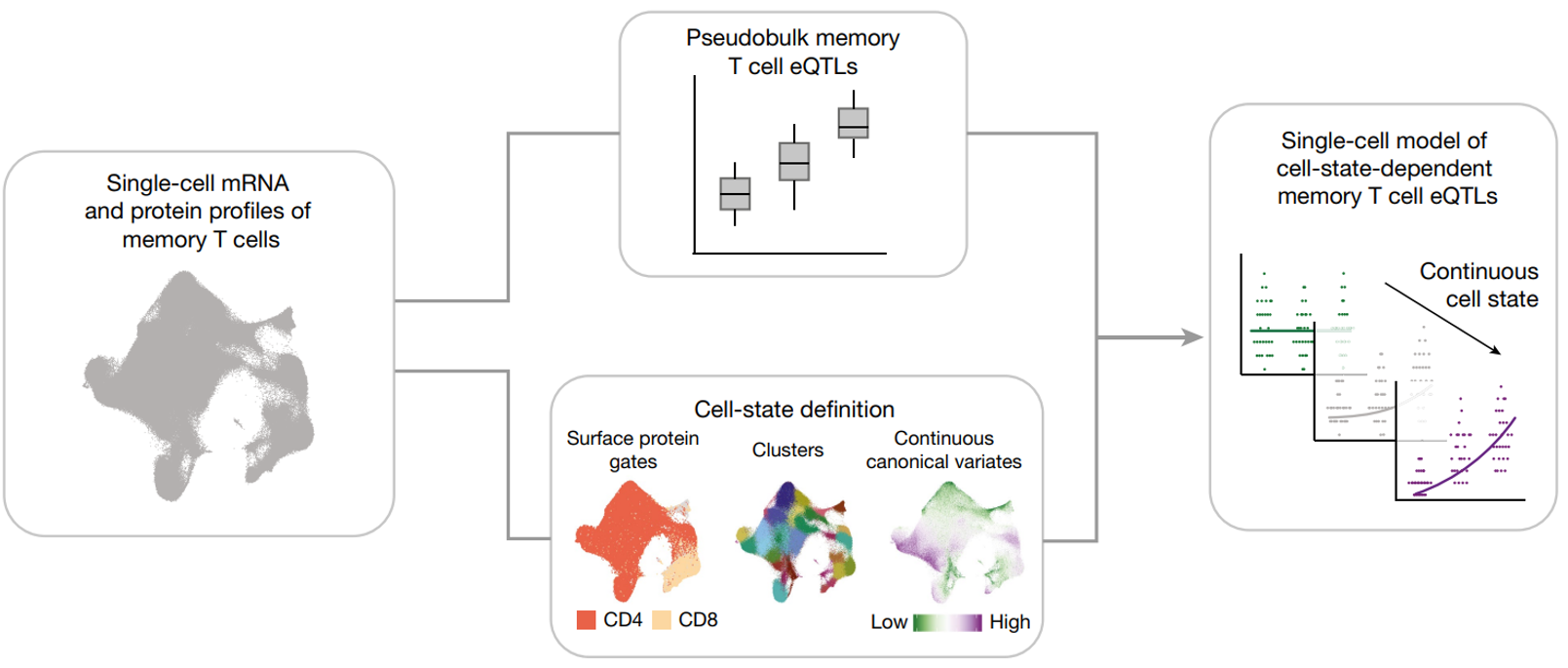
文章的分析思路如下:
-
细胞状态定义:首先,研究收集了单细胞水平的 mRNA 和蛋白质谱数据,以捕捉记忆 T 细胞的多样性。通过这些数据,定义了细胞的状态,包括表面蛋白标志物(如 CD4 和 CD8)、聚类和连续的正则变异。
-
Pseudo-bulk 记忆 T 细胞 eQTL 分析:通过将所有记忆 T 细胞的数据整合成“pseudo-bulk”数据集,定义了一组适用于所有记忆 T 细胞的稳健 eQTL。这种分析不考虑细胞的具体状态,而是识别那些在整个细胞群体中都能稳定表现的 eQTL。
-
单细胞模型的状态依赖性 eQTL 分析:最后,利用单细胞数据,进一步分析这些 eQTL 在不同细胞状态下的表现差异。特别关注连续的细胞状态,这些状态可以提供比离散的状态定义(如简单的 CD4/CD8 分类)更多的生物学信息。
挑选出顺式 eQTL (nGene)
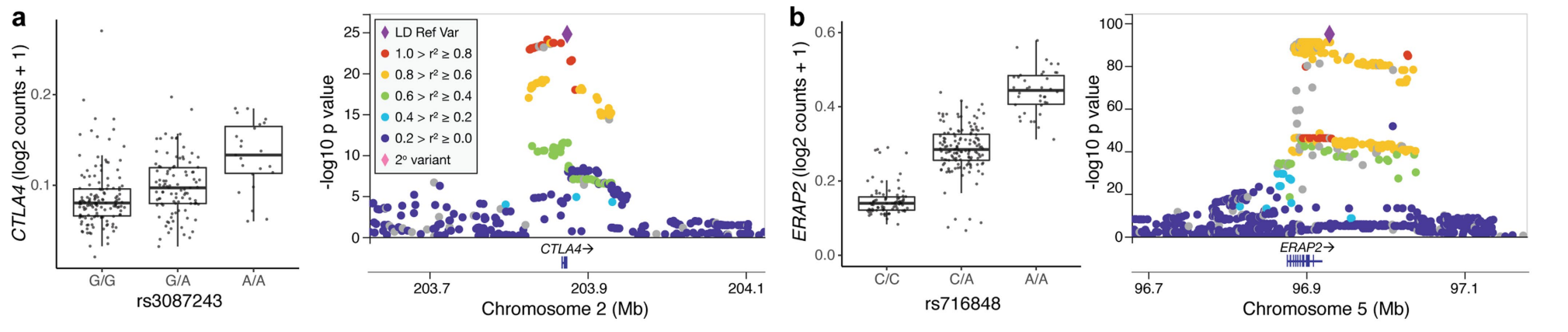
研究测试了 15,789 个基因的协变量校正表达与每个基因转录起始位点 (TSS) 不到 1 Mb 的变异之间的顺式 eQTL 关联,筛选出 6,511 个基因的顺式 eQTL(eGenes,q< 0.05),例如 CTLA4 和 ERAP2:箱线图展示了CTLA4/ERAP2基因在不同基因型下的表达量。可以看到,不同基因型下,CTLA4/ERAP2的表达量存在差异。右侧的曼哈顿图显示了在这两个基因附近的染色2区域上的eQTL关联信号,点的颜色和大小代表了连锁不平衡(LD)的不同程度和p值的显著性。
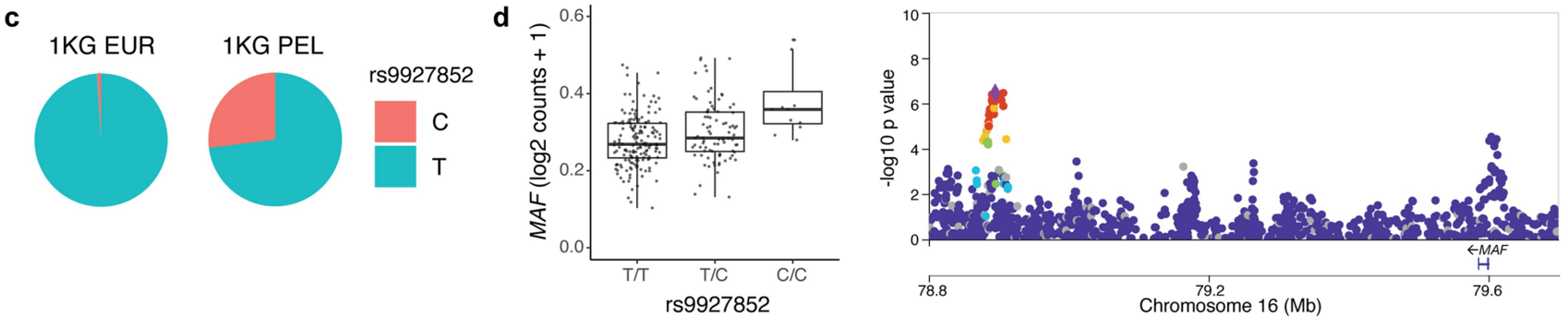
808 个 eQTL 具有可能特定于人群的影响;也就是说,由在秘鲁样本个体中常见(次要等位基因频率(MAF)> 0.05)但在欧洲个体中罕见(MAF < 0.05)的遗传变异驱动。例如,编码 MAF 转录因子的基因的 eQTL(MAF;rs9927852)在欧洲队列的(EUR) eQTL 研究中未报告,而在秘鲁队列中有(PEL)。
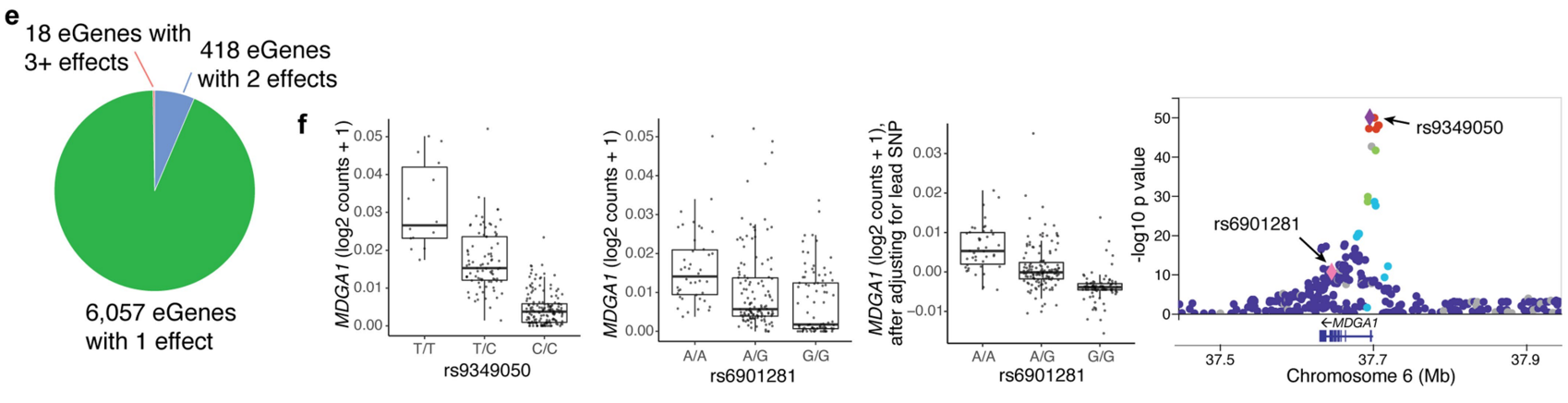
研究迭代地调节每个 eGene 的先导 eQTL (n = 6,511) 时,在 436 个位点观察到多个独立效应。饼状图显示了具有不同独立效应数量的 eGene 分布。大多数 eGene(6,057 个)只显示出一个效应,而有 418 个 eGene 具有两个独立效应,18 个 eGene 具有三个或更多效应。这表明在一些基因位点上存在多个独立的遗传变异影响基因表达。如MDGA1 附近的染色体 6 上的 eQTL 信号。rs9349050 和 rs6901281 这两个 SNPs 都显示了显著的 p 值,提示它们各自对 MDGA1 的表达有独立的影响。
:::: column
::: column-left
:::
::: column-right
:::
::::
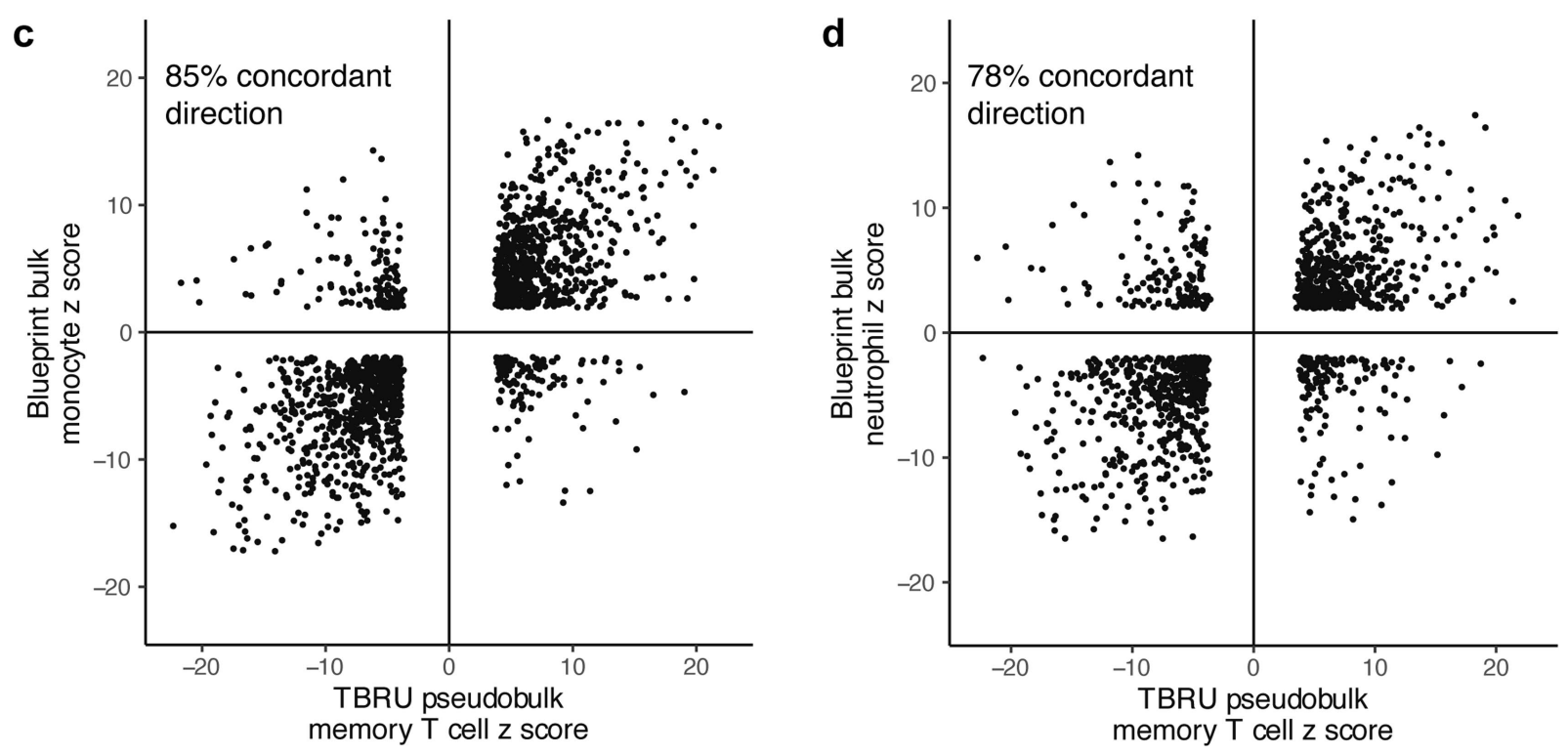
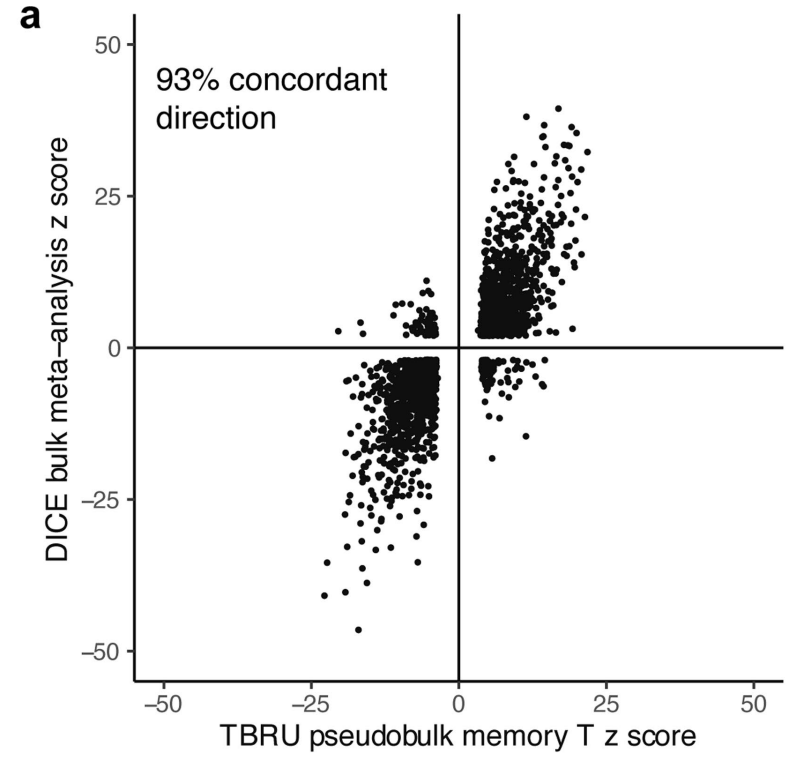
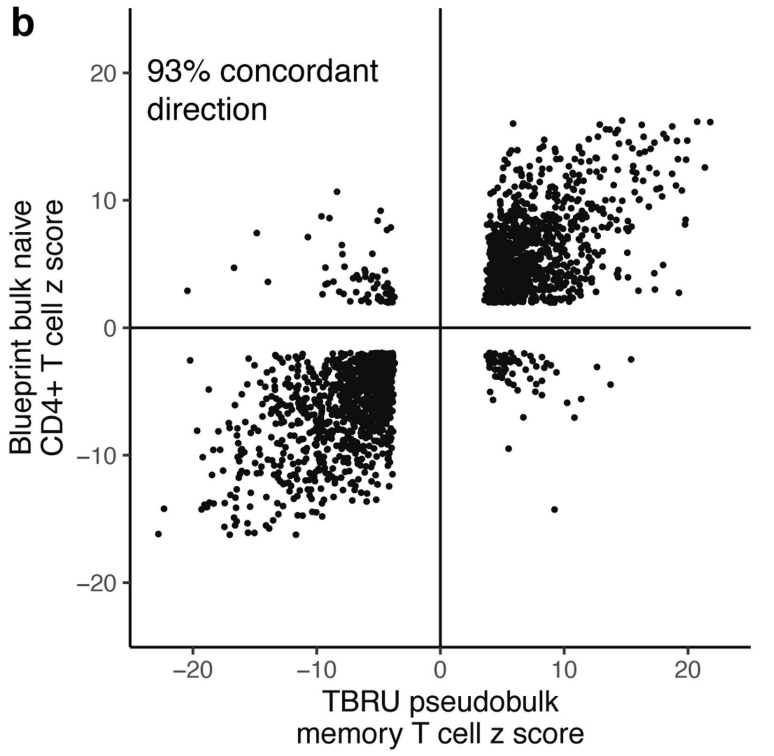
(a-b)将显著效应的变异与来自免疫细胞 eQTL 数据库 (DICE) 和 BLUEPRINT 表观基因组项目的已发表的大量记忆 CD4+ T 细胞 eQTL 进行了比较,发现 eQTL 具有高度的方向一致性。表明这些 eQTL 可能在不同细胞环境中具有一致的生物学作用。
(c-d)在 BLUEPRINT 中检测的其他细胞类型中,eQTL 与我们的记忆 T 细胞 eQTL 重叠较少,且方向一致性较低。但跨越不同功能细胞类型时有所下降,这反映出 eQTL 效应的细胞特异性特征。
2.T 细胞多样性跨越连续状态
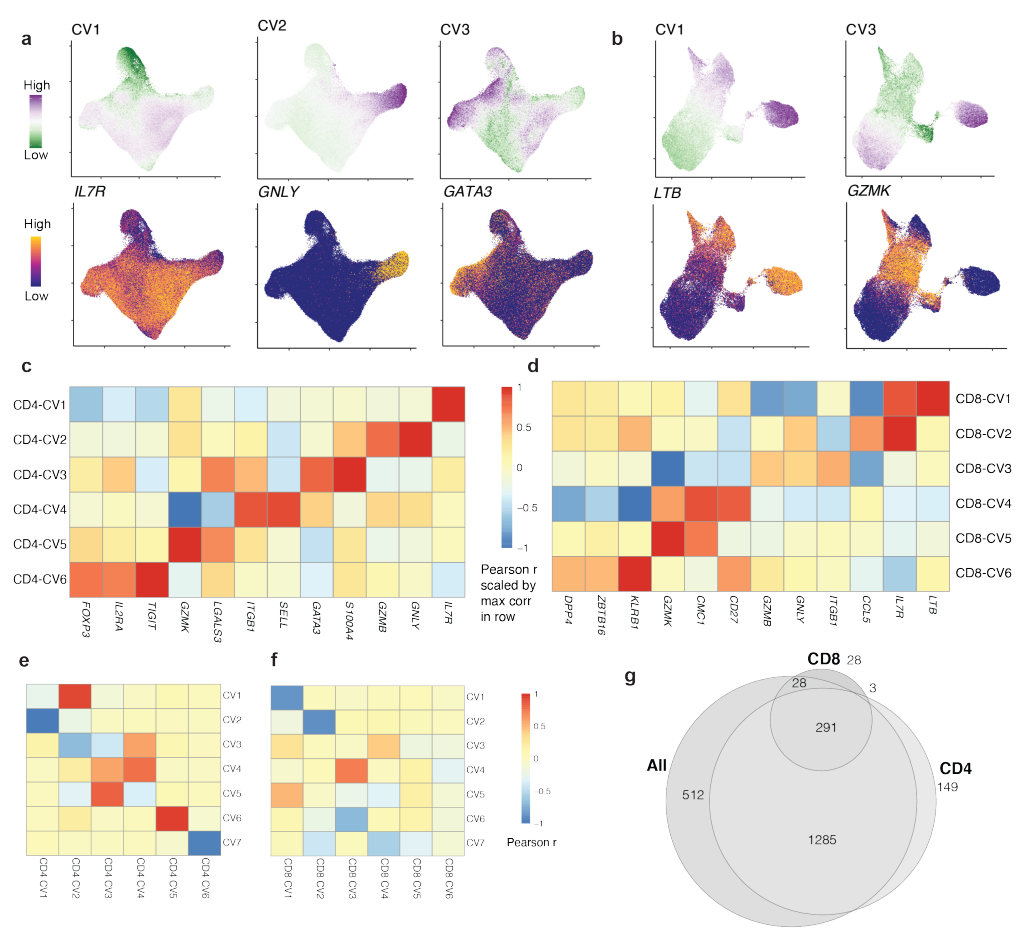
结合CITE-seq技术获取的单细胞mRNA和蛋白质测量数据,不仅允许我们对细胞进行离散聚类,还能够在多模态低维嵌入中捕捉连续状态,这些嵌入是通过典型相关分析(CCA)获得的。
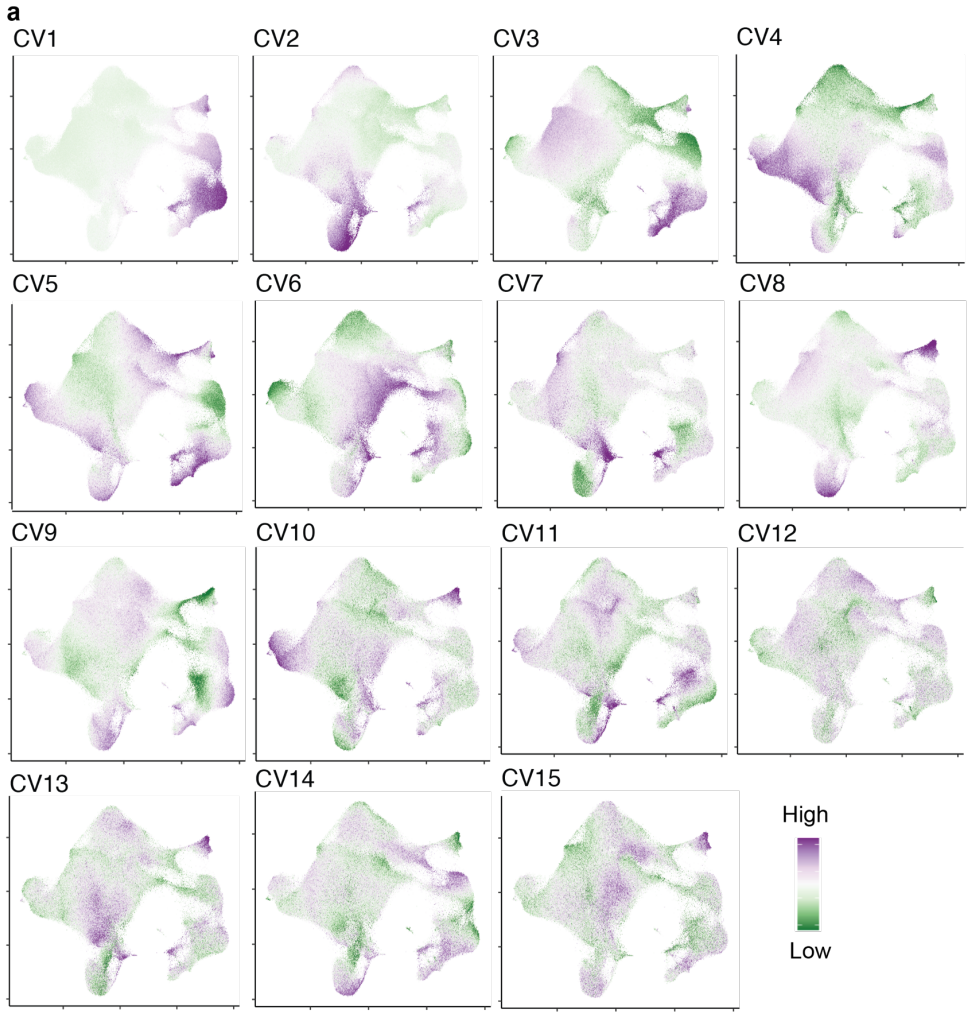
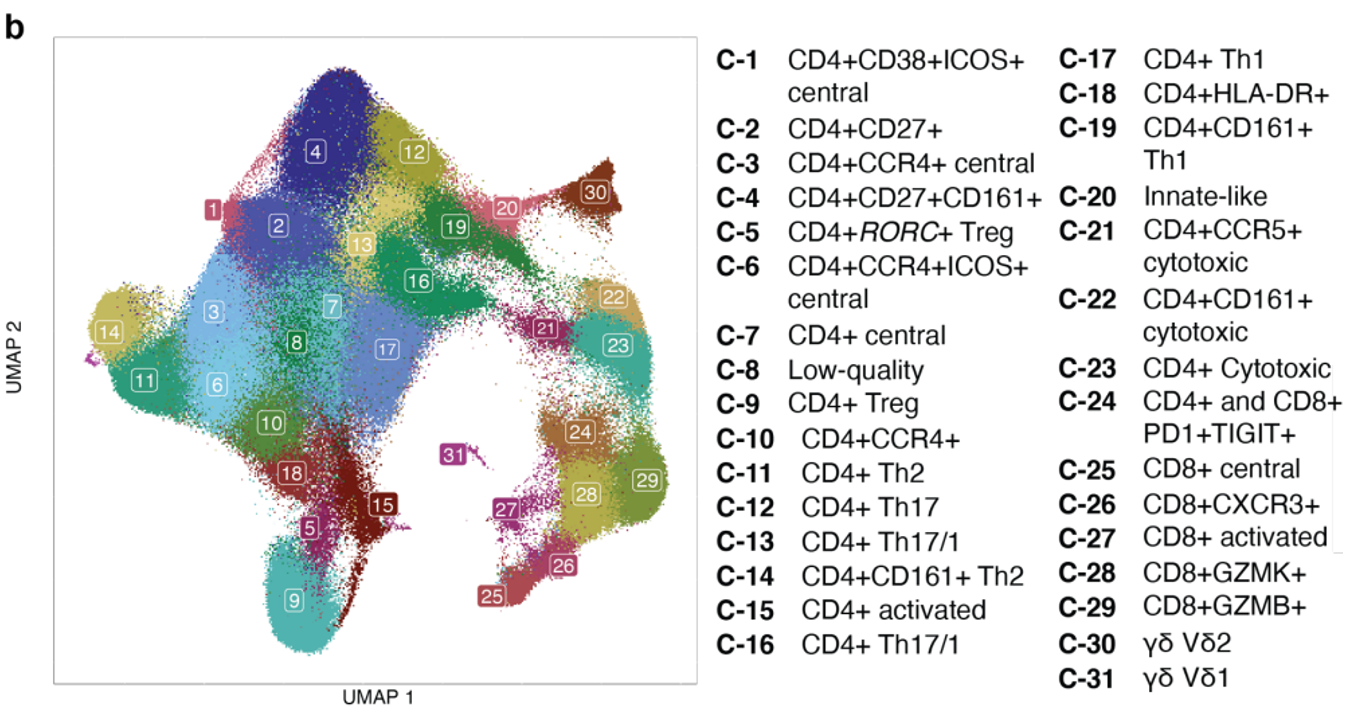
(a-b)在这些单细胞数据的原始分析中,我们基于前20个独立的CCA维度(典型变量,CVs)对细胞进行聚类,利用模态之间共享的变异来稳健定义细胞状态,并将其与T细胞标记、临床和人口统计变量相关联,而不仅仅依赖于mRNA测序数据。在这里,作者再次基于前20个CVs对每个细胞进行评分,但这次没有在CVs上进行聚类,而是将它们用作细胞状态的连续、独立表示。
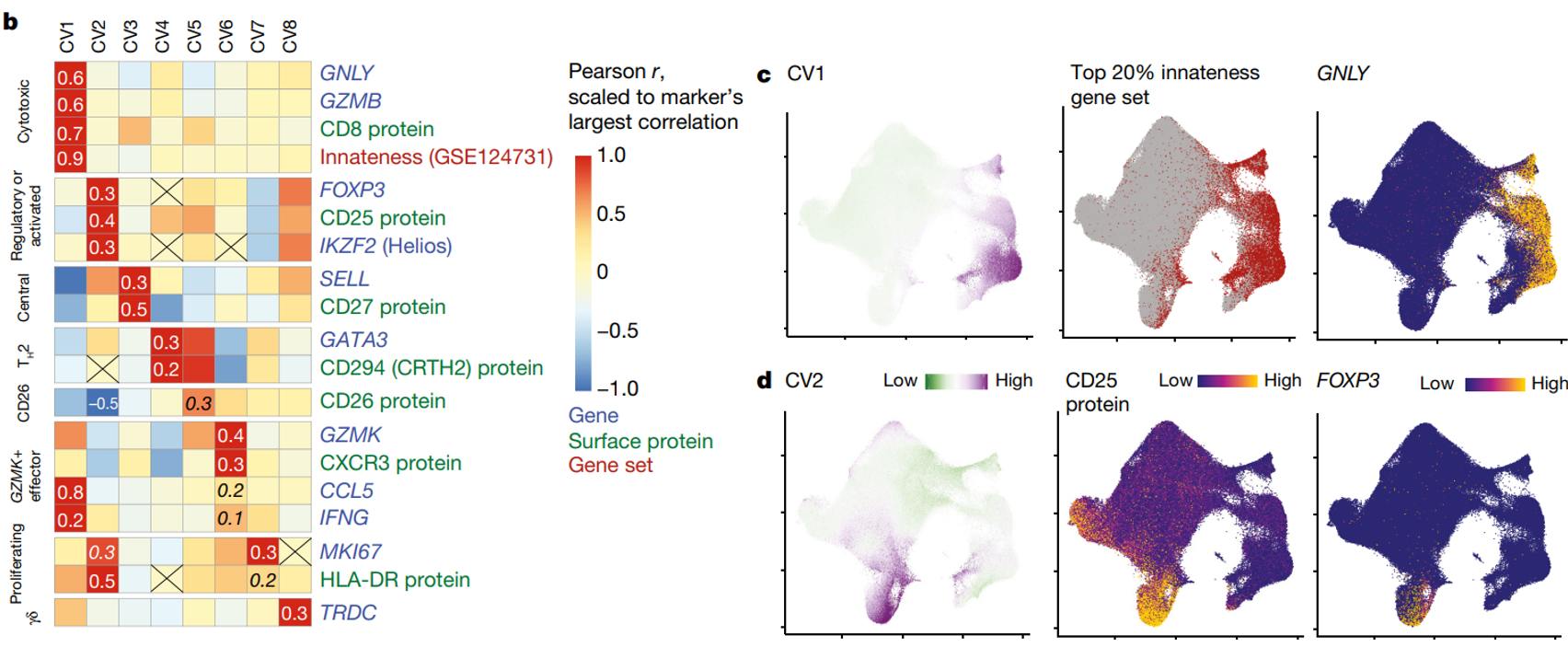
(b-d)CV 与基因、蛋白质和基因组相关,这些基因、蛋白质和基因组与描述良好的 T 细胞功能相关。
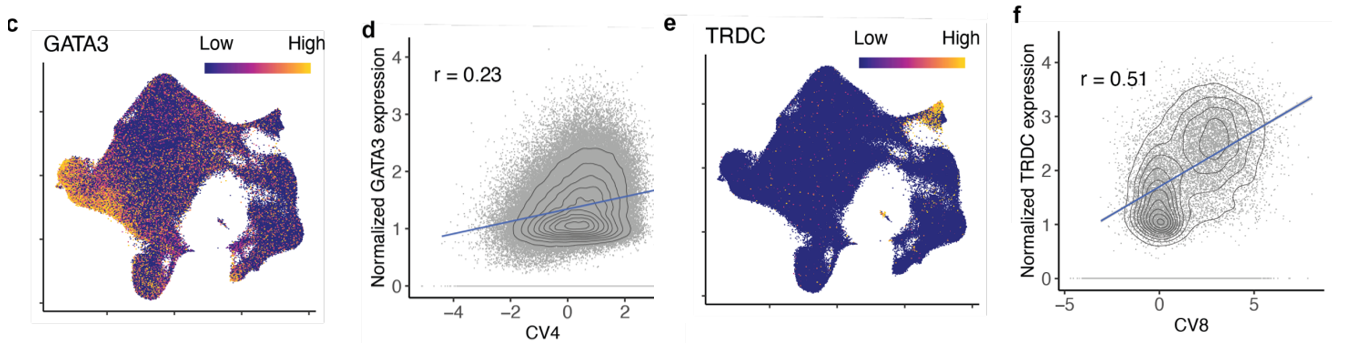
(c-f)一些 CV 与谱系定义 T 细胞状态标记相关;例如,CV4 和 TH2 的biomarker GATA3,或 CV8 和 γδ T 细胞biomarker TRDC。
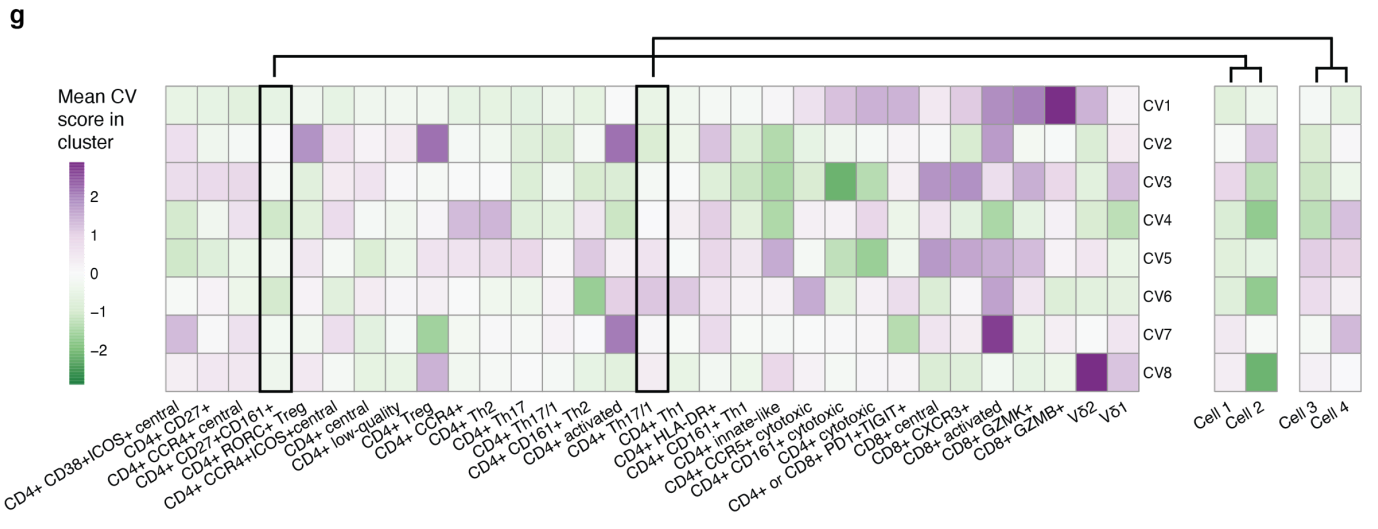
(g)热图展示了每个细胞亚型 CV(1-8)的平均得分。右侧的单独列显示两个簇中各两个细胞的 CV 得分。可见 CCA 定义的同一细胞亚型中,不同细胞的CV评分有所不同,说明CCA定义的细胞亚型掩盖了其中的细胞间异质性。因此,连续的 CV 可能更准确地表示 T 细胞的不同状态。
3.状态依赖性 eQTL 的单细胞模型
单细胞分辨率 eQTL 和 bulk eQTL 需要不同的统计模型。对单细胞使用泊松混合效应 (PME) 回归,它可以对离散和连续的单细胞状态、泊松分布的UMI count和批次结构进行建模。
基因型(整体 eQTL)和细胞状态(差异表达)效应的基线PME模型
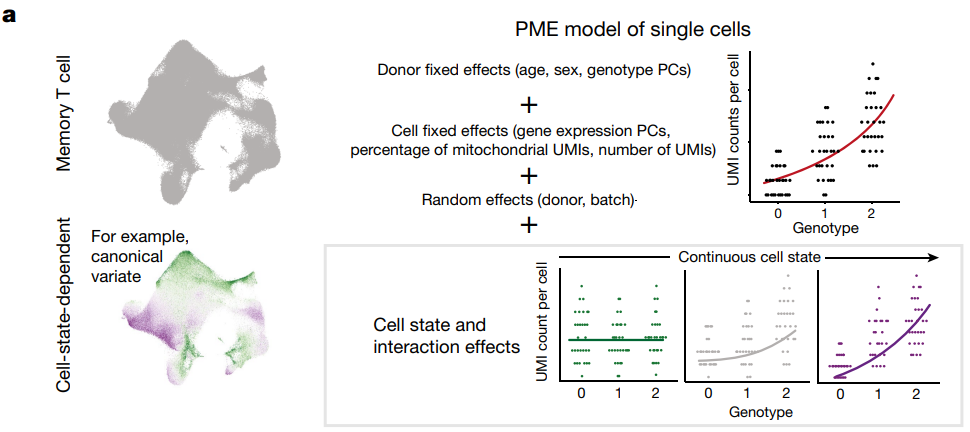
(a)将每个基因的 UMI count的泊松模型对单细胞 eQTL 进行建模,并调整常见的 eQTL 混杂因素(年龄、性别、基因型主成分 (PC) 和基因表达 PC)和在单细胞数据中显示具有基因特异性效应的协变量(UMI 计数和线粒体 UMI 百分比)。这个模型可以用于估计特定基因型在不同细胞状态下对基因表达的影响,特别是在单细胞数据的复杂背景下,有助于揭示基因表达的复杂调控机制,并与其他生物学变量相关联。
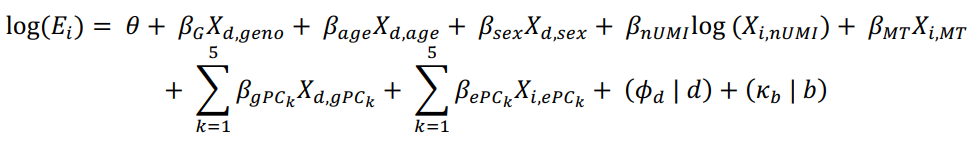
其中,E 表示第 i 个细胞中基因的表达量,
θ
\theta
θ 是截距,所有其他的
β
\beta
β 代表固定效应,如所示(nUMI = UMI的数量,MT = 线粒体UMI的比例,gPC = 基因型主成分,ePC = 单细胞mRNA表达主成分,已经进行了批次校正),这些共变量分别适用于细胞 i、供体 d 或批次 b。供体和批次被建模为随机效应截距。
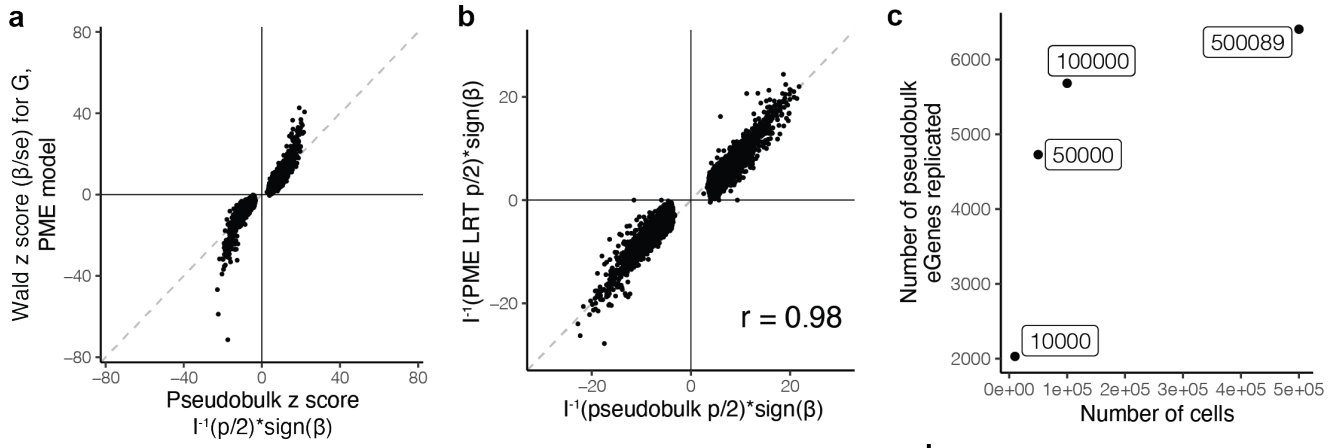
(a-c)研究使用似然比检验 (LRT) 评估了模型,并将效应大小与 Wald 统计数据进行了比较。为了证明与常用线性模型的一致性,研究用 PME 模型重新分析了数据,成功地重现了几乎所有的pseudo-bulk eQTL(q < 0.05, π 0 π_0 π0 = 0.38,6,402/6,511 = 98%),效应方向一致(6,509/6,511 = 100%)。而且随着细胞数量的增加,检测到的pseudo-bulk eGene数量也增加,这表明数据量的增加可以提高检测的可靠性。
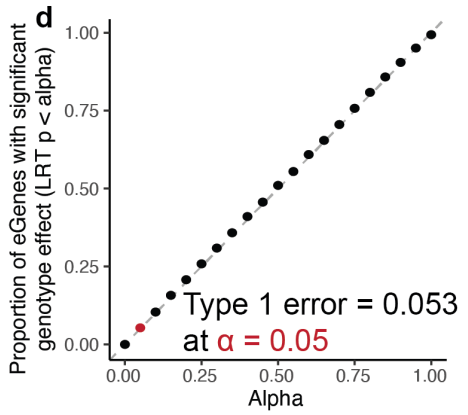
(d)展示了I型错误率(假阳性率)随着显著性水平(α值)的变化情况。横轴(α):表示显著性水平,即选择的统计显著性阈值。纵轴:表示具有显著基因型效应的eGene比例(根据LRT p值小于α的标准)。图中点的排列显示了这些eGene的显著性效应比例随着α值的增加而增加,并且基本上沿着对角线排列。这表明该方法的I型错误率与预期的显著性水平(α值)相符,表明模型在不同显著性水平下的假阳性控制良好。
在加入基因型和细胞状态相互作用项后
为了识别具有细胞状态依赖性效应的 eQTL,研究在基因型和细胞状态之间添加了一个相互作用项。
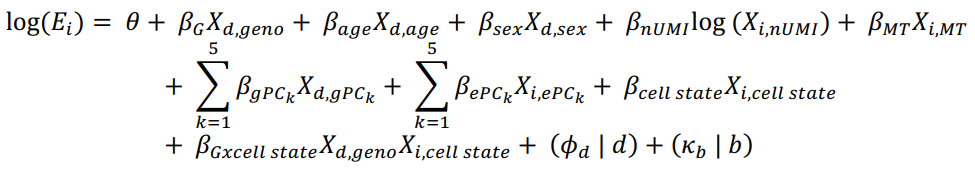
首先,在门控二元测试(CD4+ 与 CD4- )中,作者评估了相互作用模型(在所有细胞上运行)和上面验证的两个非相互作用模型(在门控 CD4+ 细胞上运行)之间的一致性——传统的pseudo-bulk线性模型、没有相互作用项的单细胞 PME 模型。
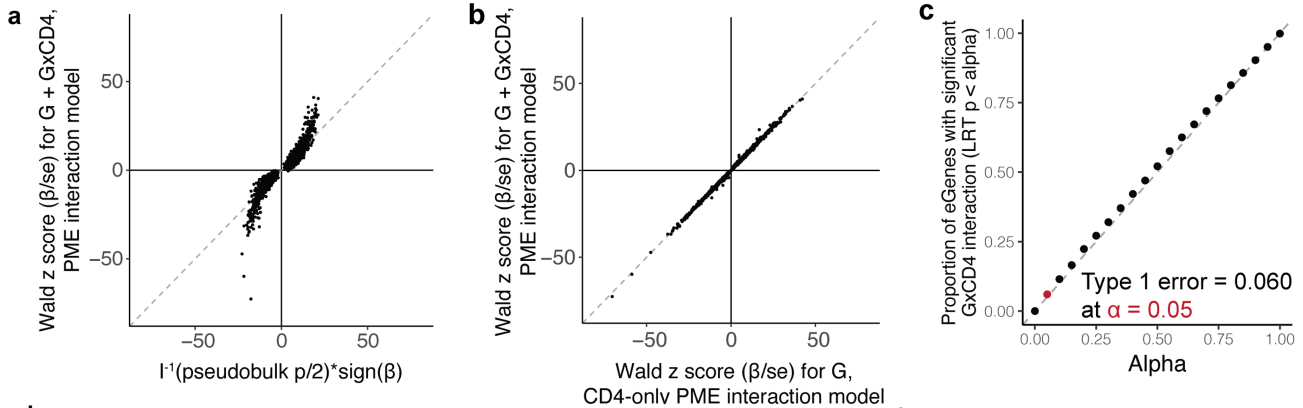
(a-c)CD4+ 细胞中总的 eQTL 效应 (βtotal = βG + βG×CD4) 与这两个模型中估计的基因型效应一致,当我们排列细胞状态时,交互项的 I 型错误在 α = 0.05 处得到很好的控制。
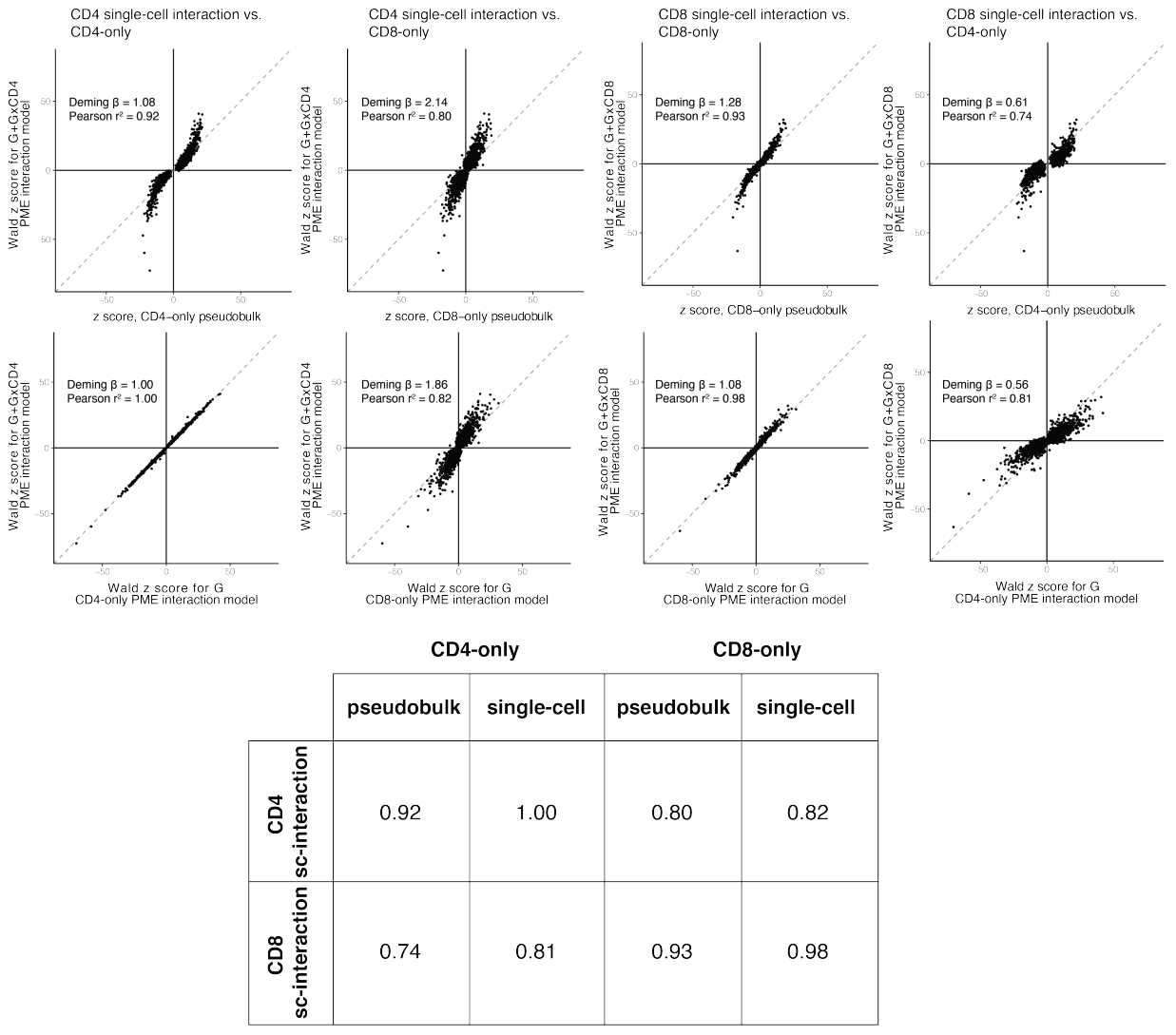
随后,研究将从相互作用模型估计的 CD4+ 和 CD8+ 依赖效应与在相同和相反谱系中测量的效应与非相互作用模型(即门控 CD4+ 或 CD8+ 记忆 T 细胞的pseudo-bulk或单细胞 eQTL 模型)进行比较。可见,CD4 β总细胞和门控CD8+ T细胞的pseudo-bulk细胞或单细胞模型之间的效应不太一致。可见有相互作用项的模型效果更不错。
标准化单细胞表达的线性混合效应 (LME) 模型
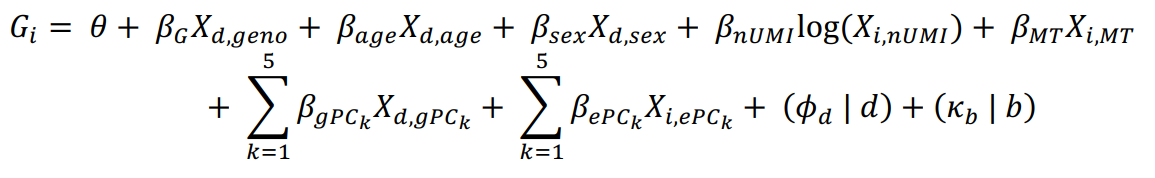

如果不考虑细胞状态,LME 的表现与 PME 相似
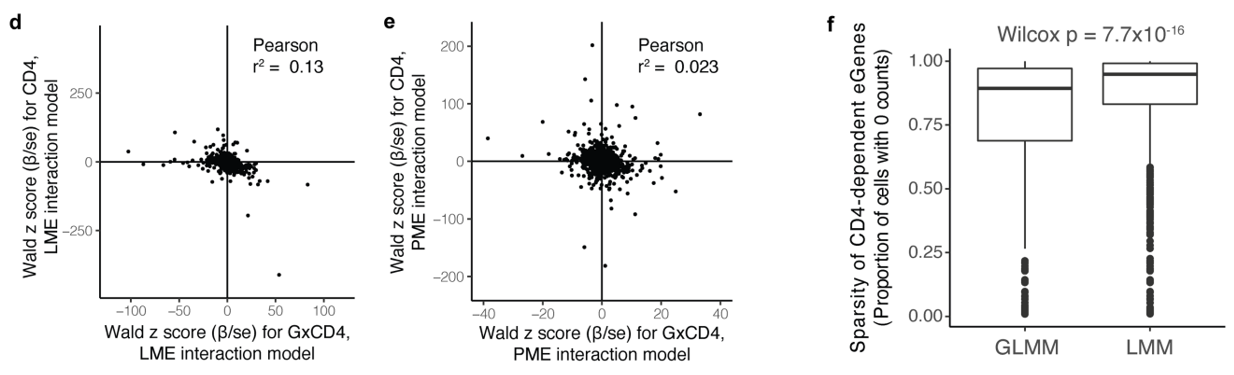
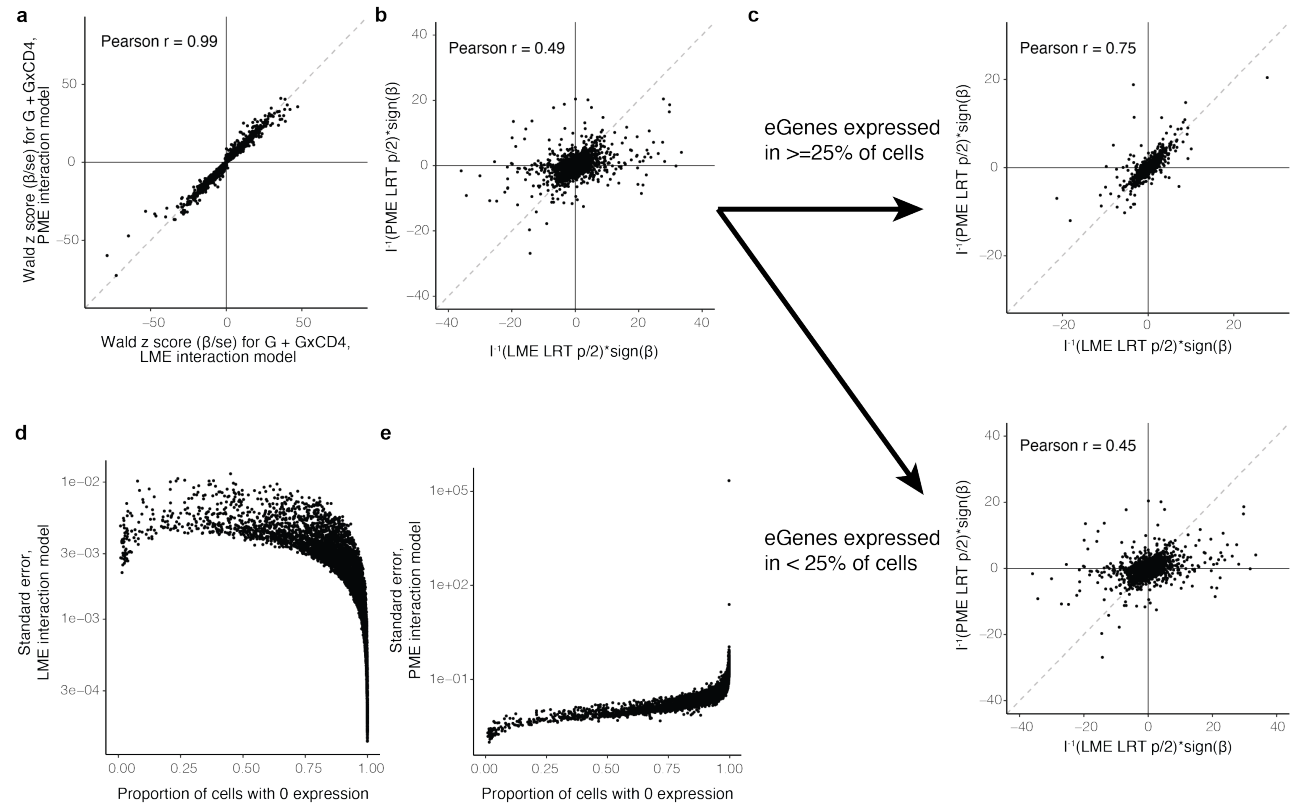
然而,如果有一个交互项,LME 会因稀疏性和细胞状态表达差异而混淆:
(上d-e)显示了LME模型和PME模型之间在检测基因效应时的一致性。LME模型中G×CD4交互作用的Wald z评分与PME模型的Wald z评分之间的Pearson相关系数非常低(r² = 0.13和0.023),可能是因为LME模型没有充分考虑到基因表达的稀疏性和细胞状态的异质性,导致了交互效应估计的偏差。
(f)展示了CD4依赖的基因在细胞中的稀疏性。稀疏性高的基因在不同的模型中可能会有不同的表现,特别是在GLMM和LMM模型中。Wilcoxon检验结果(p = 7.7×10⁻¹⁶)显示了模型之间在处理稀疏性数据时的显著差异。
(c)将基因分为在≥25%的细胞中表达和<25%的细胞中表达两类。图中展示了LME模型和PME模型之间的LRT统计量差异。在稀疏性较低(≥25%表达)的基因组中,模型间一致性较高(Pearson r = 0.75),而在稀疏性较高的基因组中(<25%表达),一致性明显降低(Pearson r = 0.45)。这表明稀疏性对LME模型的影响更大,而PME模型可能更好地处理了这种稀疏性。
(下d-e)展现了模型残差(标准化误差)与基因稀疏性的关系。在LME模型中,随着稀疏性增加,标准化误差显著增加,而在PME模型中,这种关系不明显。
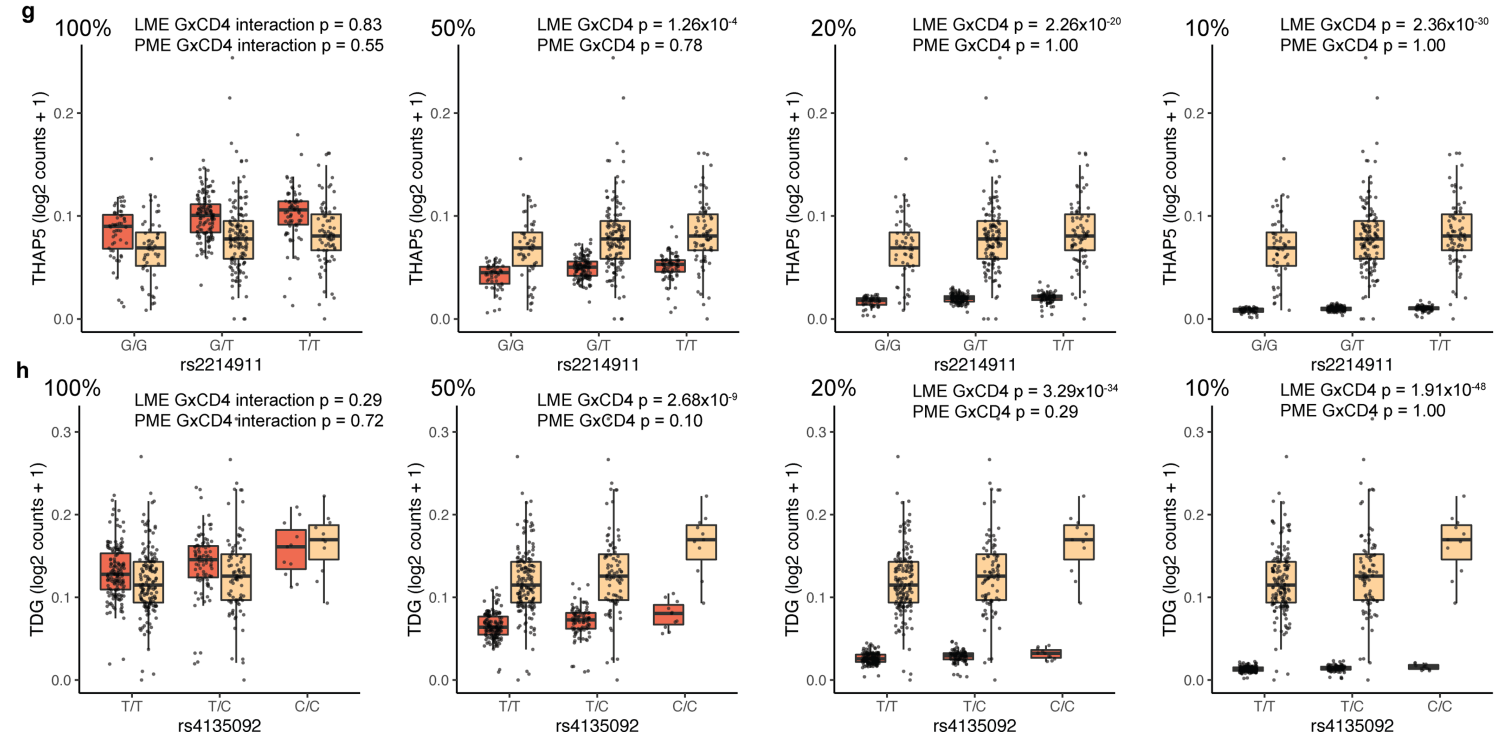
(g-h)当模拟 CD4+ 和 CD4- 细胞之间的差异表达时,LME 错误地检测到了高度显著的状态特异性 eQTL,而 PME 没有检测到。这表明LME模型不足以描述单细胞基因表达。
4.eQTL 随连续细胞状态而变化
相互作用效应独立于差异表达和主要基因型效应,并且发生在稀疏性较低的基因中
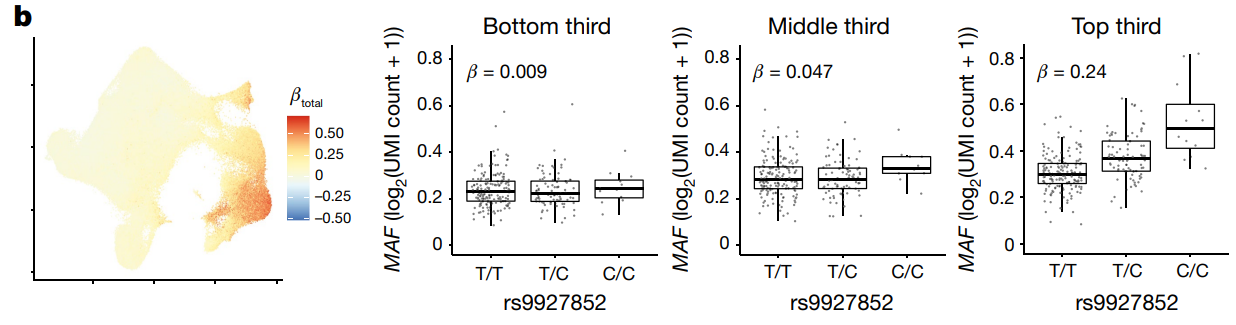
eQTL 随着细胞状态而变化。捕获细胞毒功能的 CV1 与 1,094/6,511 个记忆 T 细胞 eQTL 显着相互作用(q < 0.05)。例如,与 MAF 的 rs9927852 eQTL 的相互作用在具有较高 CV1 分数(top third)的细胞中放大了效应。
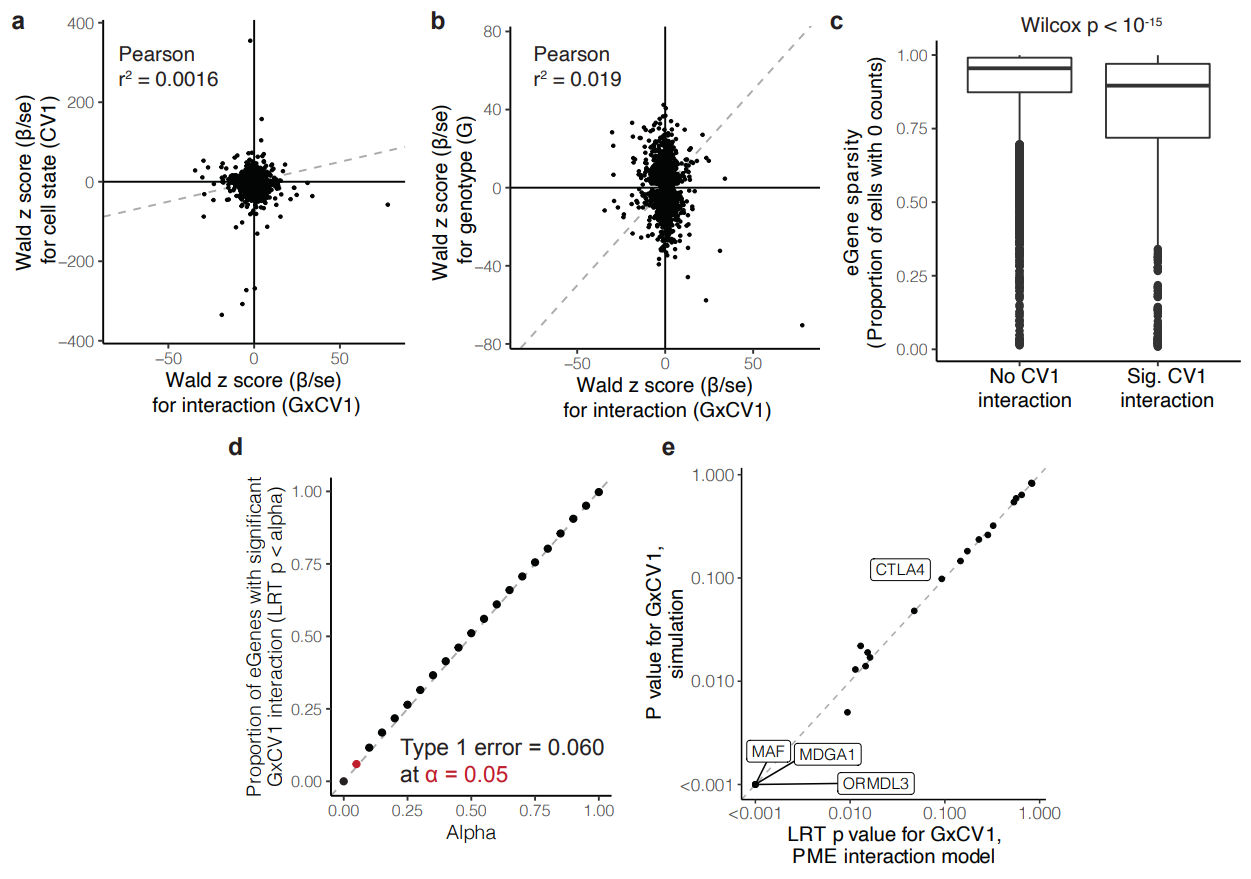
(a)表示细胞状态(CV1)的β系数(βCV1)的Wald z评分与细胞状态和基因型交互作用(βGxCV1)的Wald z评分之间的关系。这里的Pearson相关系数非常低,表示两者之间几乎没有线性相关性,意味着细胞状态本身的效应和其与基因型的交互作用效应在这些基因中是相对独立的。
(b)表示基因型(βG)的Wald z评分与细胞状态和基因型交互作用(βGxCV1)的Wald z评分之间的关系。这里的Pearson相关系数也很低,同样显示了基因型效应和其与细胞状态的交互作用效应之间的低相关性。
(c)图中比较了有显著CV1交互作用的基因(q < 0.05)(Sig. CV1 interaction)和无显著CV1交互作用(No CV1 interaction)的基因的稀疏度,后者在大多数细胞中显示零表达,表明这些基因在大部分细胞中是稀疏的。前者也显示出较高的稀疏度,但略低于无显著交互作用的基因,表明这些基因在细胞中的表达更为分散。说明CV1的交互作用效应可能与基因表达的稀疏度有关。
(d)显示了不同α值下,检测到具有显著GxCV1交互作用的基因比例。可见 I 型错误得到很好的控制。
(e)比较了模拟数据中和实际PME交互作用模型中对于GxCV1交互作用的LRT p值。可见,模拟数据和实际数据之间的结果高度一致。图中标记了几个基因(例如CTLA4, MAF, MDGA1, ORMDL3),这些基因在实际数据和模拟数据中均显示显著的GxCV1交互作用。
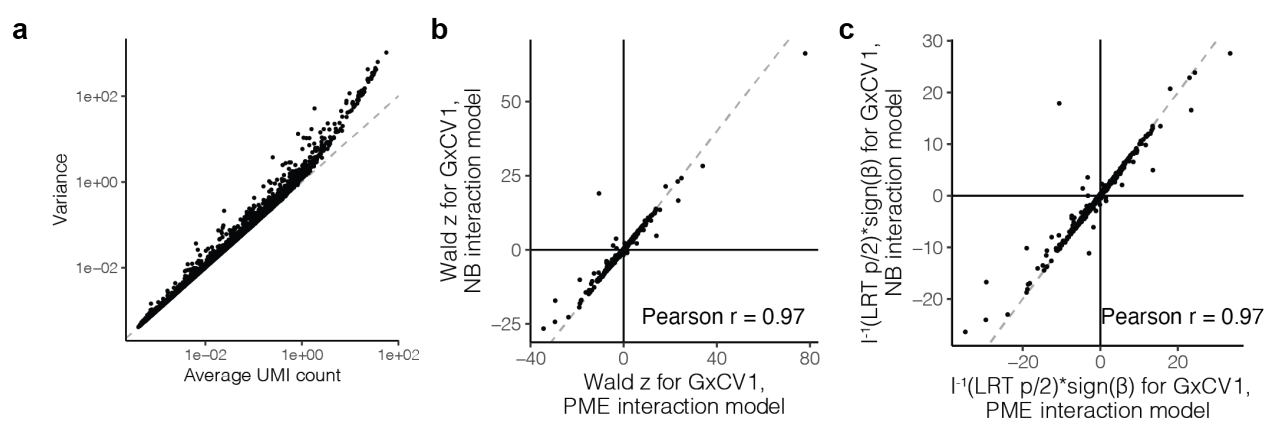
泊松和负二项模型的相互作用对于测试的 1,000 个 eGenes 子集基本一致,基因表达大多没有过度分散。
连续细胞状态比离散表型捕获了更多状态依赖性调节变异信息
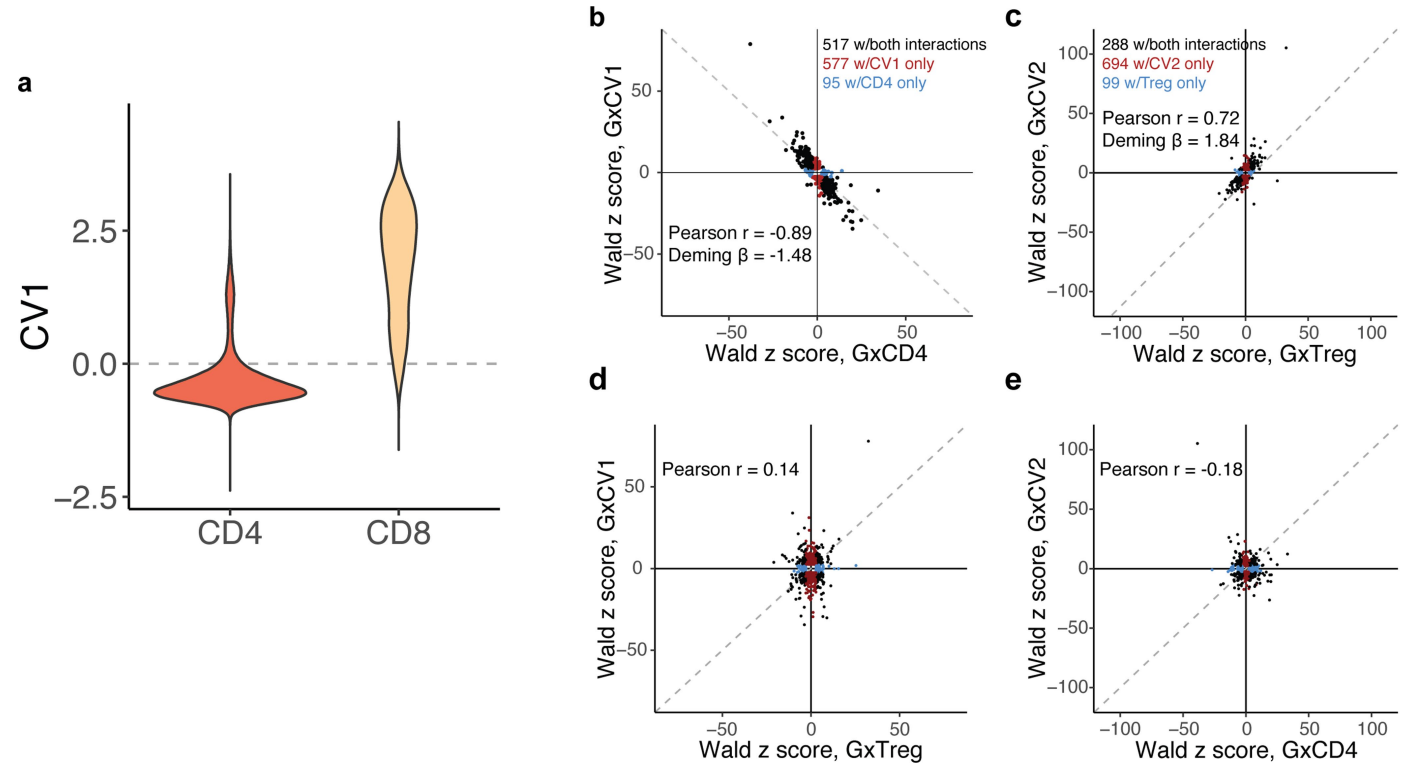
(a)展示了CD4+和CD8+ T细胞在CV1分数上的分布。可见CV1能够区分这两种细胞类型,CV1分数小于0的主要是CD4+细胞,而CV1分数大于0的主要是CD8+细胞。
(b)展示了GxCV1的Wald z评分与GxCD4的Wald z评分之间的关系。图中显示,有517个eGenes在这两种交互作用中均显著,577个eGenes仅在CV1交互中显著,而95个eGenes仅在CD4+交互中显著。Pearson相关系数(r = -0.89)和Deming回归斜率(β = -1.48)表明,虽然方向一致,但CV1相互作用通常更强。
(c)展示了GxCV2的Wald z评分与基因型和Treg细胞(GxTreg)的交互作用之间的关系。288个eGenes在两者中均显著,694个仅在CV2交互中显著,而99个仅在Treg交互中显著,前者明显超过了后者。Pearson相关系数(r = 0.72)和Deming回归斜率(β = 1.84)也支持了CV2的交互作用在检测eQTL中的作用。
(d-e)展示了CV1和GxTreg,CV2和GxCD4之间的交互作用的特有性。这说明CV1和CV2可以捕捉到在离散状态下被掩盖的调控变异。表明在单细胞数据中捕捉连续状态可以更好地理解生物学调控过程。
连续细胞状态也可以更好地解释许多异质性 eQTL 效应
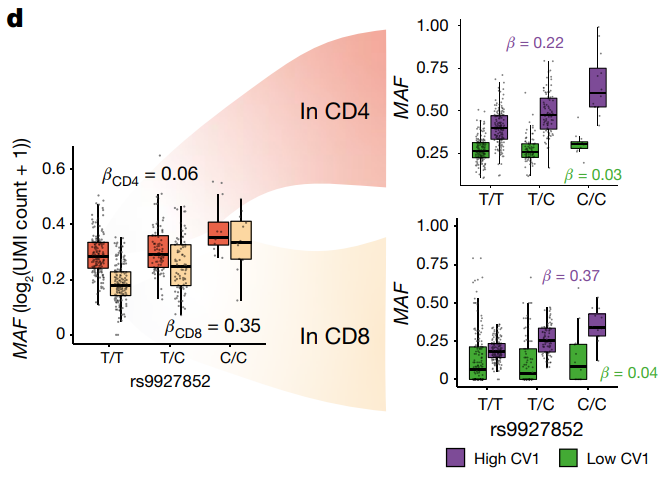
MAF eQTL 在 CV1 得分高的 CD4+ 和 CD8+ 记忆 T 细胞中都很强(βCD4,高 CV1 = 0.22,βCD8,高 CV1 = 0.37),但在 CV1 得分低的细胞中较弱(βCD4,低 CV1 = 0.03,βCD8,低 CV1 = 0.04)。在联合模型中,对于在两个单变量模型中相互作用均显著的 264/517 eQTL,CV1 相互作用的显著性同样取代了离散 CD4+ 相互作用,而对于 49/517 eQTL,CD4 相互作用取代了连续相互作用。因此,尽管一些 eQTL 可能是由谱系驱动的,但观察到的大部分调控变异最好通过细胞毒性程度来解释。
5.单个细胞具有不同的 eQTL 效应
为了捕捉单个细胞中多面状态的调控效应,研究将正交 CV 依次添加到多变量 PME eQTL 模型中。
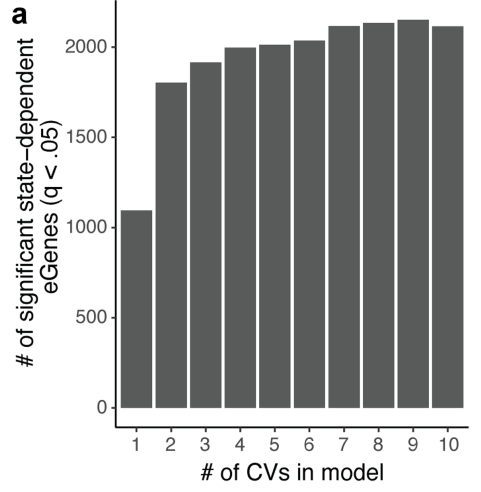
显著相互作用的 eGenes 数量在 7 个 CV 时达到稳定水平,在单变量模型中,8 个及以上的 CV 识别的 eQTL 相互作用比早期的 CV 少。
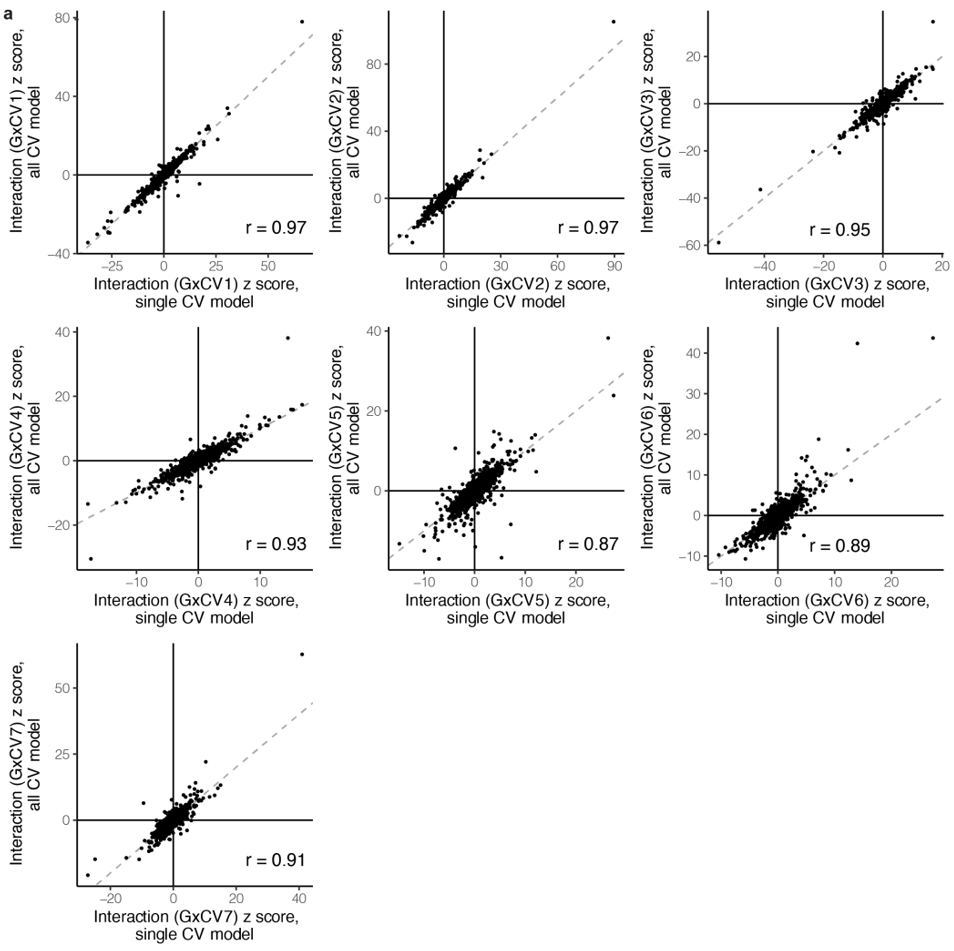
(a)研究在多变量模型中纳入了 7 个 CV,并观察到与单变量相互作用的高度一致性(r = 0.87-0.97),与正交 CV 的独立性一致。
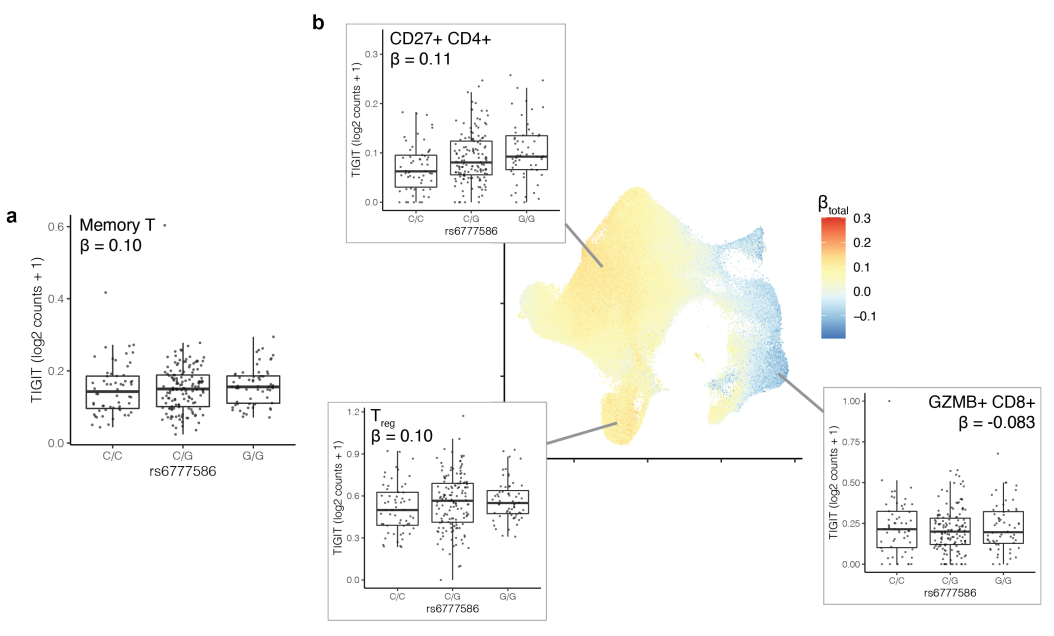
一些非pseudo-bulk显著性eQTL依然表现出状态依赖性(390/8,692,q < 0.05),表明这些基因的表达和特定状态(如细胞类型或亚型)有关,例如 TIGIT。图a展示了记忆T细胞中的TIGIT基因表达与基因型rs6777586的关系,发现β值为0.10,表示轻微的表达差异。图b进一步细分显示了CD27+ CD4+ T细胞、调节性T细胞(Treg)和GZMB+ CD8+ T细胞中TIGIT基因的表达。各个图中的β值(如CD27+ CD4+中的β=0.11,Treg中的β=0.10,GZMB+ CD8+中的β=-0.083)表明不同亚群中eQTL效应的差异。
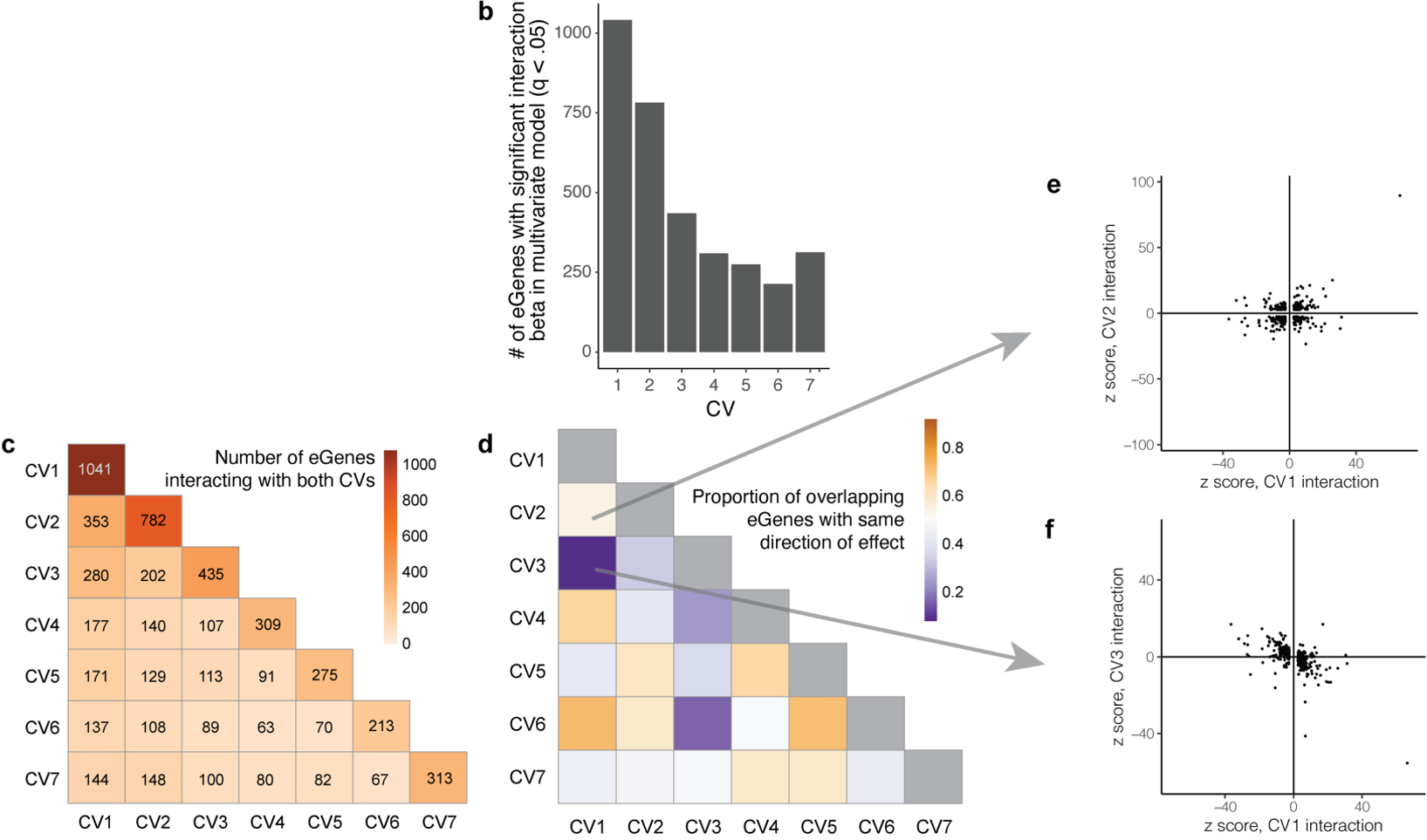
(b)CV1 在单变量和 7-CV 多变量模型中都具有最多的相互作用 eGenes。
(c-d)一些 eGenes 与多种细胞状态显着相互作用,c图展示了不同CVs之间共同作用的eGenes数量,数字越大,表示两个CV之间有更多共同作用的eGenes;d图展示了不同CVs之间具有相同作用方向的eGenes比例。
(e-f)在不同CVs(CV1与CV2、CV1与CV3)之间eGenes的z分数比较。图中显示出一些基因在不同CVs之间的相互作用方向一致,如CV1(细胞毒性)和CV6(TH1),而在另一些情况下方向相反(如CV1和CV3)。通过这种多层次分析,可以更好地理解不同细胞状态下基因调控的复杂性和多样性。
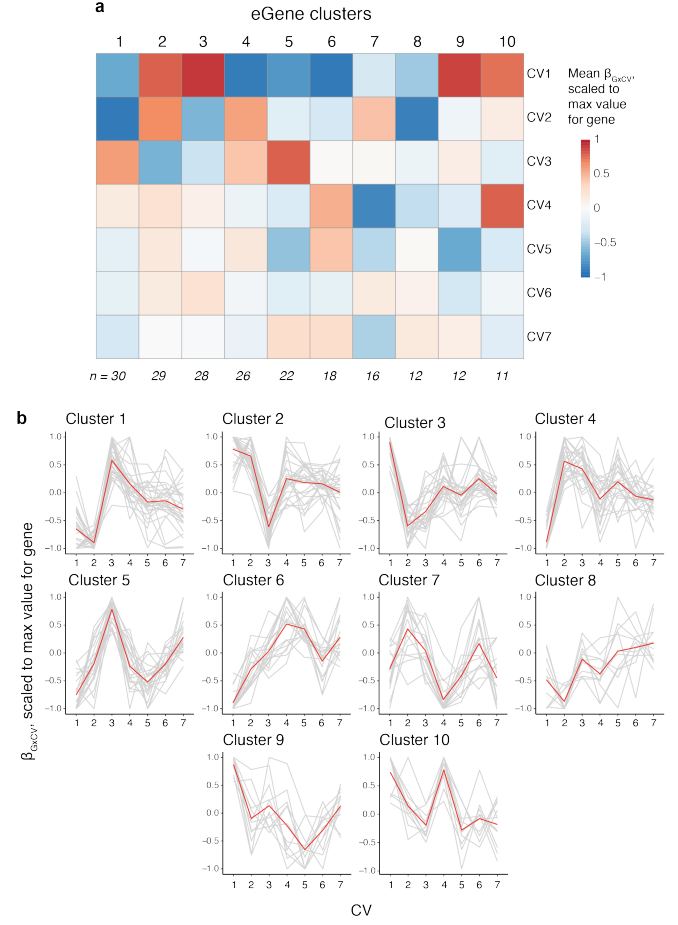
根据基因的缩放多变量相互作用 β 值(相对于主效应的方向)对其进行聚类,定义了 10 种广泛的 CV 相互作用模式,这些模式可能反映了共同的细胞状态依赖性调节机制。
b线图揭示了每个聚类中基因的交互作用模式,可以看到不同聚类中基因对CV的响应是不同的。有些聚类对特定CV反应显著,而其他聚类则表现出不同的响应模式。使用 HOMER,观察到一些转录因子结合基序在与每个 CV 相互作用的 eGenes 启动子中富集,或与相互作用的lead 变异重叠,包括已知的 T 细胞转录因子,如 RUNX1。很少有如此显著的富集,这表明调控环境可能比单个转录因子驱动程序更复杂。
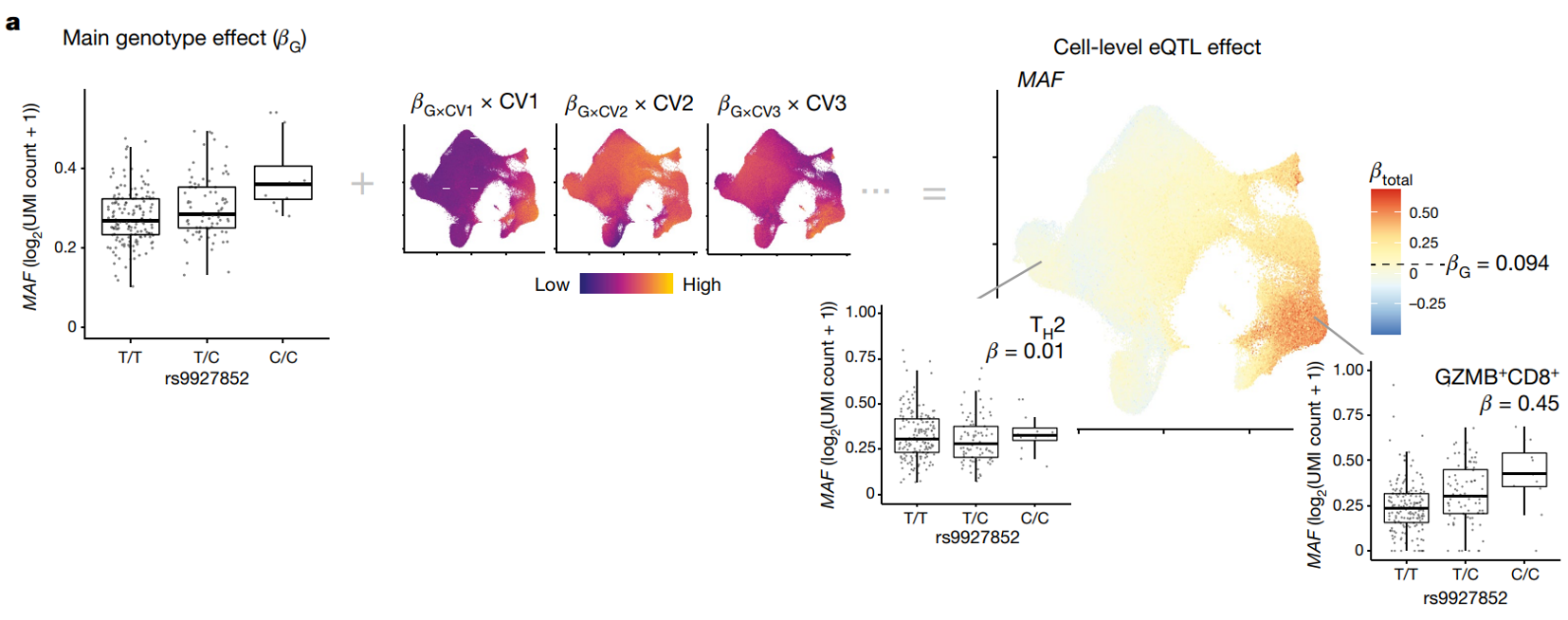
研究通过将相互作用 β 值与每个细胞对应的 CV 分数相加,以单细胞分辨率估计 eQTL 效应。这些 CV 分数捕捉了可能调节调节活动的每个状态的部分影响。将此值添加到基线基因型 β 可估计总细胞水平 eQTL 效应,该效应在细胞间变化,与 eGene 表达无关。
6.eGenes可以产生多种动态效应
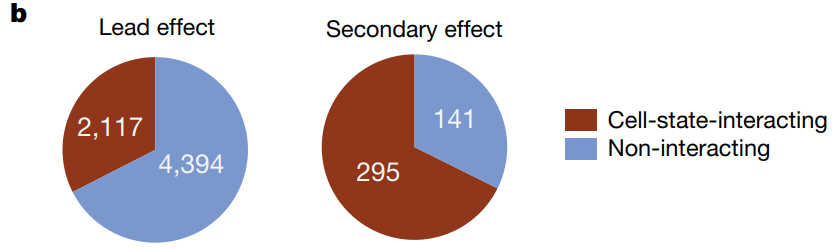
先前的研究表明,以先导效应为条件的次级 eQTL 更可能是细胞状态特异性的。本研究观察到 68% 的次级 eQTL 具有显著的细胞状态相互作用,而先导变体只有 33%。
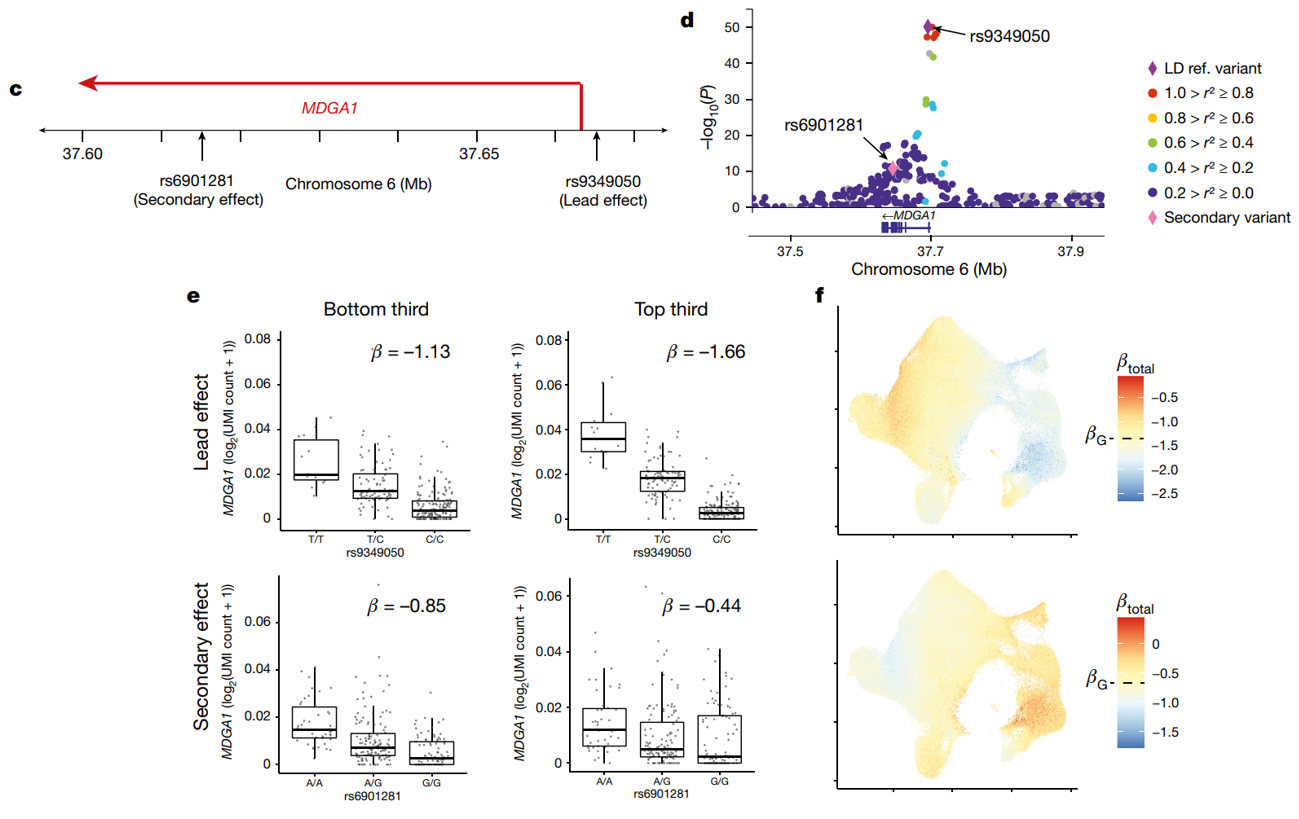
总共 203 个 eGene 具有至少 2 个独立的状态相互作用效应——有时具有矛盾的 CV 相互作用。例如,MDGA1 的先导 eQTL 随 CV1 增加,而其次级效应则减少
(c)显示了MDGA1基因在6号染色体上的位置,以及与之相关的两个变体:rs9349050(先导效应)和rs6901281(次级效应)。
(d)是MDGA1位点的详细局部放大图,展现先导变体(rs9349050)和次级变体(rs6901281)的连锁不平衡程度(r²值)。rs9349050作为主要变体具有最高的-log10§值,表明其与MDGA1表达之间的关联最为显著。次要变体rs6901281也显示出较高的显著性,表明它对MDGA1的表达也有独立的影响。
(e)展示了MDGA1在不同CV1评分下的表达变化:
-
先导效应(Lead Effect):(上左)显示了rs9349050在CV1评分最低的三分之一细胞中的基因型效应。β值=-1.13表示在这些细胞中,基因型变异对MDGA1表达有负向效应。(上右)显示了相同变异在CV1评分最高的三分之一细胞中的效应。β值=-1.66,表明变异对MDGA1表达的负向效应更强。
-
次级效应(Secondary Effect):(下左)显示了rs6901281在CV1评分最低的三分之一细胞中的基因型效应。β值=-0.85,表明次级变异对MDGA1表达的负向效应。(下右)显示了相同变异在CV1评分最高的三分之一细胞中的效应。β值=-0.44,表明次级变异的效应减弱。
(f)UMAP图显示了MDGA1的总eQTL效应在不同细胞中的分布。上部图对应于先导变体rs9349050,下部图对应于次级变体rs6901281。突显了变异体对基因表达的调控在细胞群体中的复杂性。这种可视化揭示了eQTL效应如何在不同细胞类型或状态中表现出空间异质性。
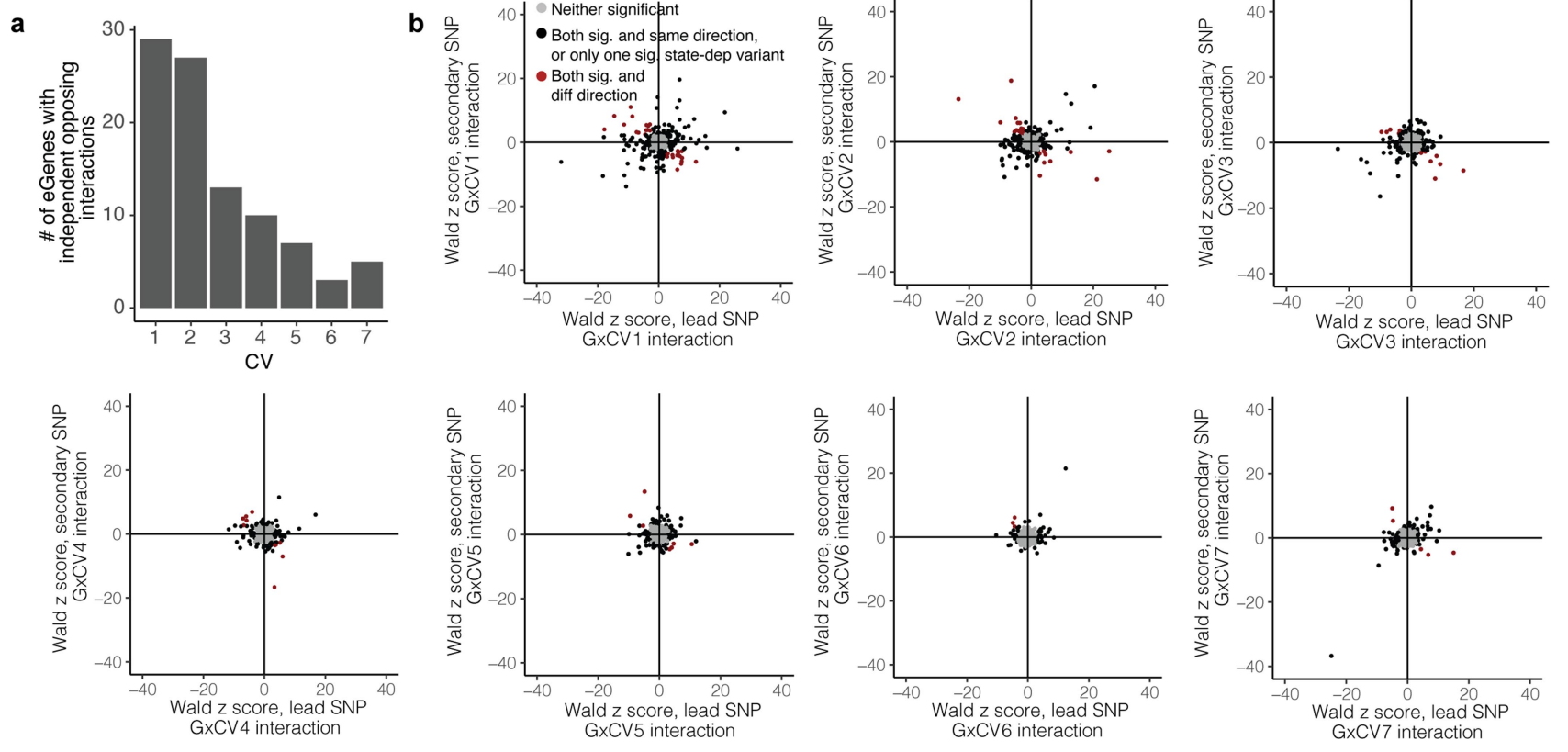
在具有至少 2 个独立 CV1 依赖效应(q < 0.05)的 60 个 eGene 中,29 个 eGene 对其先导变体和次级变体具有不同的 CV1 相互作用方向。 70 个 eGenes 显示与至少一个 CV 存在不一致。
7.许多自身免疫基因位点是动态 eQTL
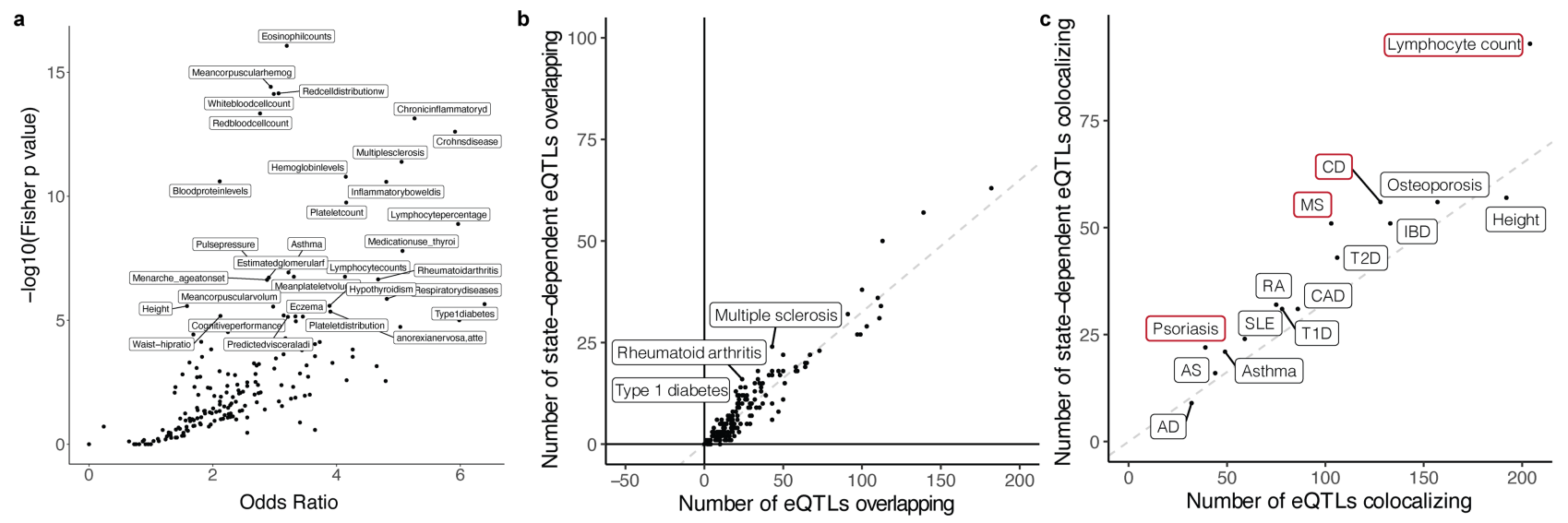
(a)横轴是比值比(Odds Ratio),表示特定性状与记忆T细胞eQTL共定位的GWAS变异的比例相对于其他性状的比例;纵轴是-log10(Fisher p值),表示性状与eQTL共定位的显著性。数值越高,显著性越强。每个点代表一种性状,如嗜酸性粒细胞计数、红细胞分布宽度和慢性炎症性疾病显示出较高的比值比和显著性,表示这些性状与记忆T细胞eQTL有较强的共定位关联。
(b)横轴是与eQTL重叠的GWAS变体总数;纵轴是与状态依赖性eQTL重叠的GWAS变体数量。每个点代表一种性状,标注的性状(如多发性硬化症、类风湿性关节炎、1型糖尿病)在这两个方面均有显著富集。
(c)横轴是与eQTL共定位的GWAS变体总数;纵轴是与状态依赖性eQTL共定位的GWAS变体数量。每个点代表一种性状,红色勾勒出的性状(如牛皮癣、AD=特应性皮炎、MS=多发性硬化症等)表示这些性状在状态依赖性eQTL中有显著富集。没有主要免疫病因的特征没有因与状态依赖性 eQTL 共定位而富集,而一些已知 T 细胞介导疾病的 GWAS 基因座,如牛皮癣和多发性硬化症与状态依赖性 eQTL 共定位的比例显着高于预期。
总的来说,了不同性状的GWAS变异在记忆T细胞eQTL和状态依赖性eQTL中的分布情况。显著富集的性状(如多发性硬化症、类风湿性关节炎、1型糖尿病等)表明这些性状可能受到特定遗传变异的调控,而这些变异在特定细胞状态下(如记忆T细胞)显现出调控作用。
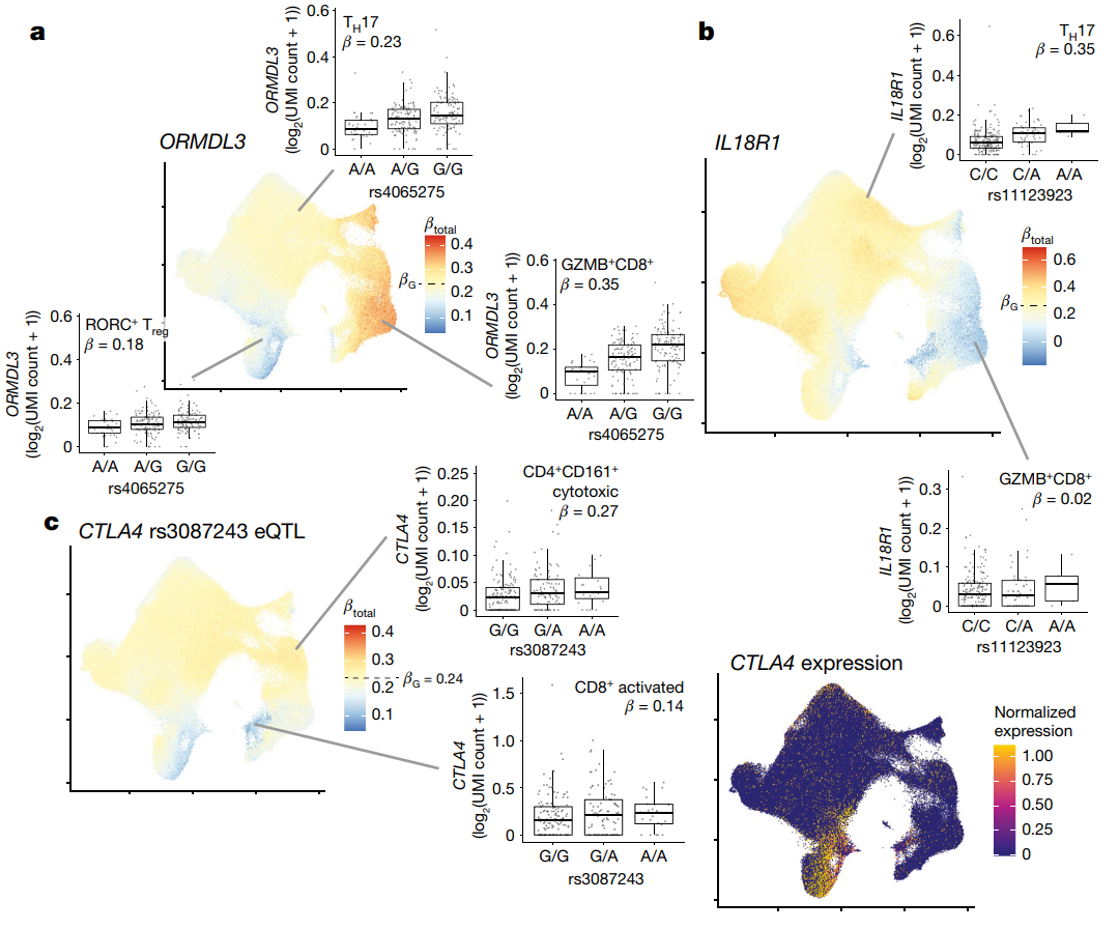
ORMDL3(rs4065275)的主要 eQTL 变体与 RA GWAS 变体(rs59716545)处于 LD(1000 Genomes PEL,r 2 = 0.69;1000 Genomes EUR,r 2 = 0.68),并与 CV 1 和 2 具有显着的多变量相互作用。
(a)ORMDL3 eQTL 在 GZMB+ 细胞毒性 CD8+ T 细胞中最强。
(b)另一方面,领先的 IL18R1 eQTL 变体 (rs11123923, chr2_102351384_C_A) — 与炎症性肠病 GWAS 变体 rs1420098 LD 一致(1000 Genomes PEL 和 EUR 中的 r 2 = 1.00)— 在 TH2 和 TH17 细胞中效果最强,在细胞毒性状态下效果较弱。
(c)GWAS 变体在总体表达较高的状态下并不总是具有更强的 eQTL 效应。例如,CTLA4 的领先 eQTL 效应由 rs3087243 (chr2_203874196_G_A) 介导,这与 RA 和 1 型糖尿病有关43。尽管 CTLA4 在 Treg 细胞、RORC+ Treg 细胞和活化 T 细胞中的表达最高,但这些细胞的 eQTL 效应较弱。
eQTL 效应在细胞毒性 CD4+ T 细胞中最强,而细胞毒性 CD4+ T 细胞的 CTLA4 表达非常低。表明疾病过程可能出现异常状态,即致病变异调节低水平基因表达。
8.动态 eQTL 位于调控区域
状态依赖性 eQTL 可能集中在调控区域,包括启动子(跨状态共享)或增强子(状态特异性)。研究基于pseudo-bulk总统计数据,对每个基因座的 eQTL 效应进行了精细映射,并在相关区域中识别了因果变异(CAVIAR)。
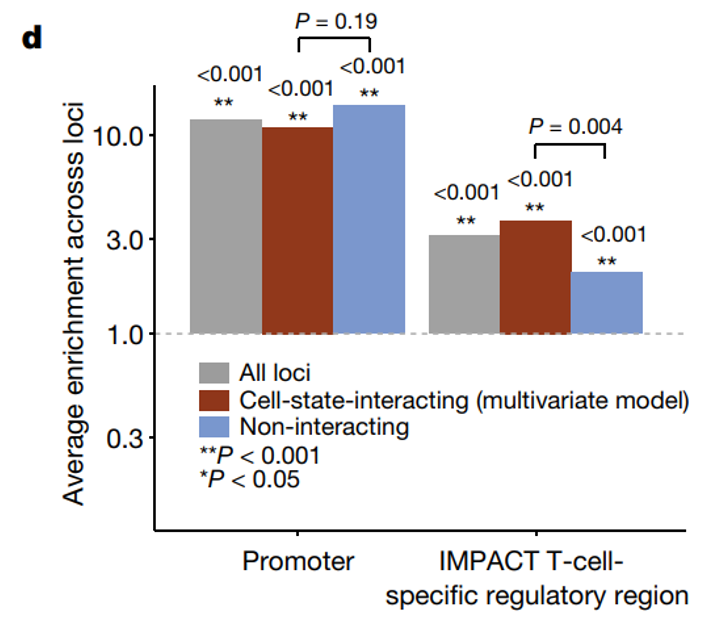
对于研究将主要效应精细映射到一个变异的基因座(n = 461,后验包含概率 (PIP) ≥ 0.5),研究计算出启动子中 eQTL 变异的 PIP 加权富集率为 12.05 倍(TSS ± 2 kb;排列 P < 0.001)。根据 7-CV 多变量模型定义的细胞状态相互作用和非相互作用 eQTL 分别富集了 11.00 倍和 14.15 倍,反映了启动子的状态无关调控重要性。
为了测试增强子的富集情况,研究使用了 CD4+ TH1 细胞中 T-bet 的表型相关活性转录 (IMPACT) 模型的推断和建模,以根据转录因子结合和表观遗传特征定义细胞类型特异性调控区。我们排除了上面定义的启动子。在 T 细胞特异性调控区域中,细胞状态相互作用 eQTL 的富集度几乎是非相互作用 eQTL 的两倍。与非相互作用 eQTL 相比,这些状态依赖性 eQTL 在 B 细胞或单核细胞特异性 调控区域中的过度表达并不显著。
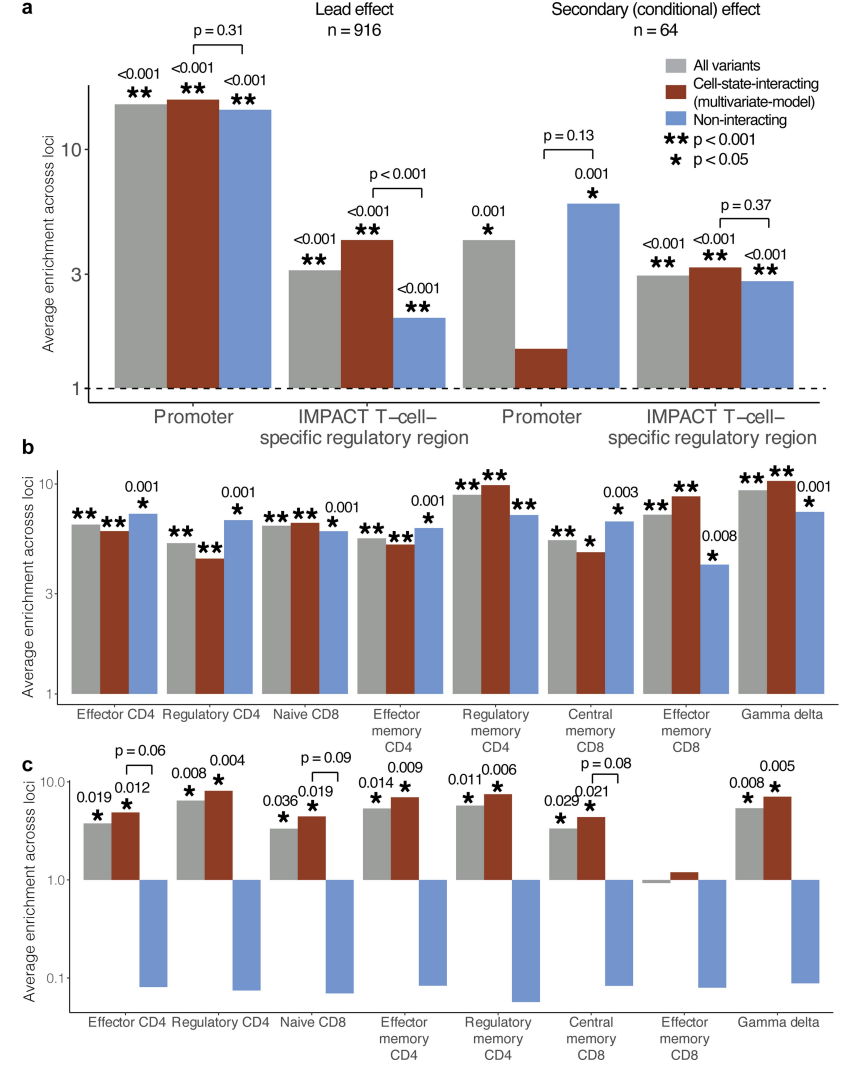
(a)为了更准确地识别因果变异,研究将这个秘鲁数据集与来自 BLUEPRINT 的欧洲数据相结合,并对pseudo-bulk效应进行了多祖先精细定位。研究对 916 个 eGenes 的先导效应进行了精细映射,将其归类为单个因果变异 (PIP ≥ 0.5)。与秘鲁分析一样,这些变异在启动子中富集 ,与非相互作用 eQTL相比,状态相互作用 eQTL 在 T 细胞特异性区域中富集程度更高。
(b)使用在 CD4 和 CD8 亚群中测定的测序 (ATAC-seq) 峰,在转座酶可及染色质测定中,状态相互作用和非相互作用 eQTL 均高度富集,这反映了启动子和远端调控区域的组合。
(c)次级 eQTL 变体及其主要的状态依赖性子集也在 T 细胞 ATAC-seq 峰中富集。
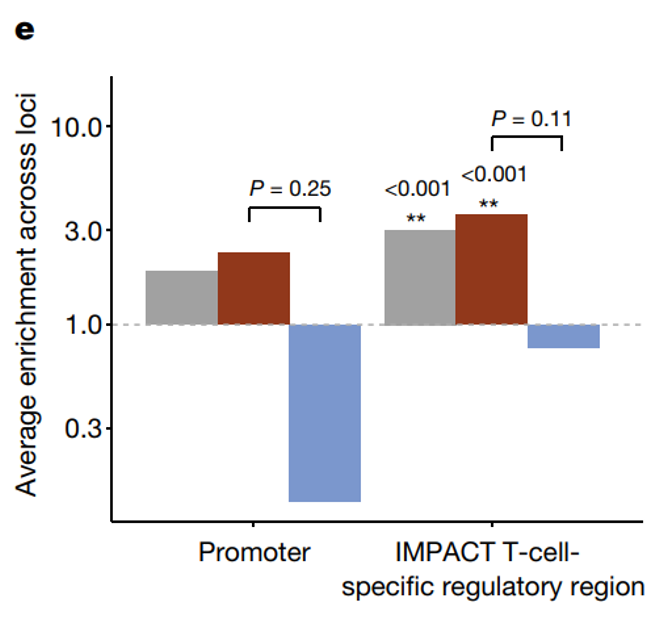
研究发现增强子中次级 eQTL 变体的表达量高于启动子,次级 eQTL 变体在启动子中的富集程度低于主要变体。它们在 T 细胞特异性调控区中的显著富集,尤其是状态相互作用的次级变体,表明具有细胞类型特异性的调控作用。
9.建模其他动态 eQTL 景观
为了评估该方法在混合细胞类型中的实用性,研究将该模型应用于已发表的外周血单核细胞 (PBMC) (n = 89) 单细胞 RNA 测序数据集,其中 mRNA PC 划定了离散的细胞类型。发现962 个pseudo-bulk eQTL 中约有三分之一沿着前 6 个 mRNA PC 变化,并与相应的离散细胞类型具有一致的相互作用。
相反,研究还通过对记忆 T 细胞数据集中 CD4 和 CD8 T 细胞内定义的 CV 进行建模,确定了 eQTL 与较大细胞类型子集内更细粒度细胞状态的相互作用。
10.eQTL与疾病状态的相互作用
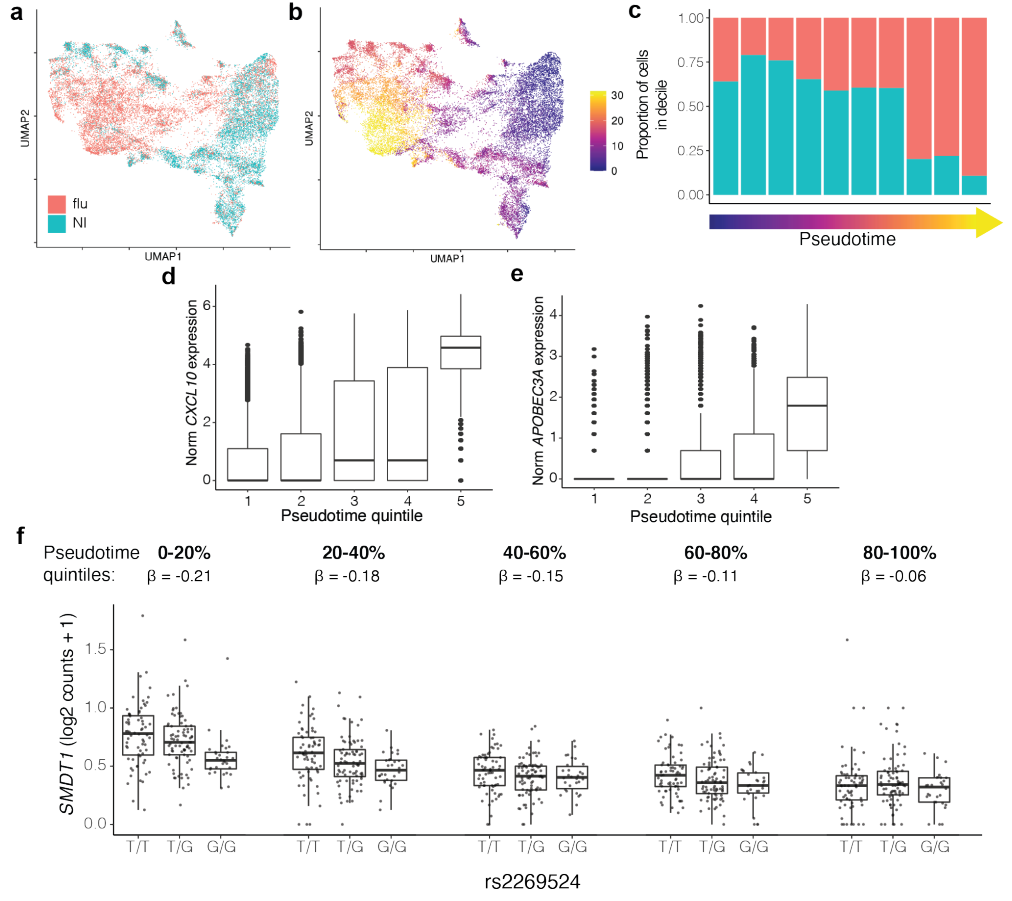
细胞状态也可以通过对扰动或分化过程中采集的细胞以及伴随的基因调控变化进行非线性轨迹或速度分析来定义。研究证明了 PME 能够模拟 eQTL 与单核细胞非线性拟时序的相互作用,这些单核细胞对甲型流感病毒或模拟感染的反应各不相同。
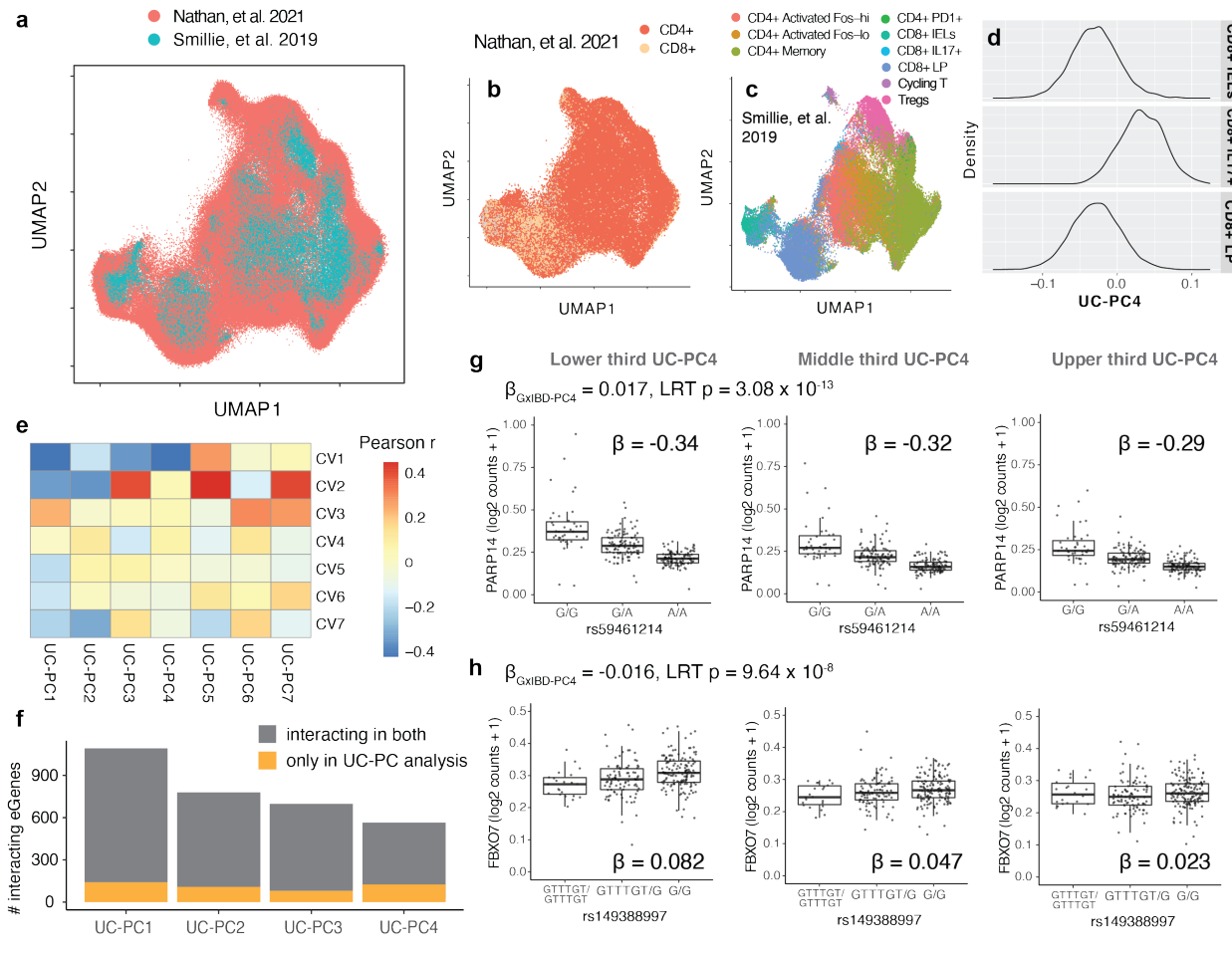
(a-d)除了体外扰动外,体内疾病状态也可能与 eQTL 相关。然而,由于患者样本稀缺,疾病分析单细胞数据集可能包含相对较少的细胞。作者建议将大型、基因分型的单细胞数据集投影到较小的疾病参考数据集上,并使用参考低维嵌入中的细胞投影作为细胞状态来测试 eQTL 相互作用。这使研究能够利用非疾病数据集的大量细胞和疾病参考的临床相关框架。通过将 500,089 个记忆性 T 细胞投射到约 70,000 个结肠 T 细胞的溃疡性结肠炎 (UC) 参考上证明了这种方法的可行性,并测试了 eQTL 与 UC-PCs4 的相互作用 。
(e-h)除了重现原始记忆性 T 细胞分析中的许多相互作用外,研究还观察到一些 UC-PCs 独有的相互作用,这些相互作用定义了致病细胞状态,可能反映了组织或疾病特异性动态 。
总结
1.单细胞数据的优势:
单细胞数据揭示了状态依赖的调控异质性,这是传统大规模方法难以捕捉的。研究强调了将eQTL分析聚焦于单细胞分辨率的紧迫性,因为传统的汇总分析可能会掩盖调控多样性。
2.PME模型的应用:
研究使用单细胞泊松模型(PME模型)更精确地定义了连续状态依赖的eQTL。这种模型利用大规模的细胞计数,提高了统计检测的能力,并解决了之前研究中常见的稀疏性问题。
通过分析单细胞中的eQTL效应,研究发现即使在同一个细胞群体内,基因表达调控效应也会因状态的不同而变化,这揭示了疾病相关变异的上下文依赖性。
3.研究实例与未来方向:
例如,RA相关的rs4065275对ORMDL3表达的影响在GZMB+细胞毒性CD8+ T细胞中最强,但更广泛地驱动了细胞毒性。独立eQTL位点中具有相反状态依赖效应的情况,表明一个位点内的状态特异性调控元件可能决定了这些调控相互作用。
4.未来挑战和方向:
随着越来越多的来自非欧洲遗传血统和不同临床表型的基因型分型单细胞数据集出现,将展示出超出已定义的细胞类型的异质性。为了在大规模基因组范围内有效地应用PME模型,需要开发更快速的广义线性混合模型,并解决多重检验问题。
5.单细胞研究的广泛应用:
连续状态不仅存在于T细胞,还适用于神经元、星形胶质细胞的空间梯度、上皮-间充质转化、成纤维细胞中的Notch信号等。在异质PBMCs中进行分析,可能需要在定义连续状态之前对个体细胞类型进行分选,或者使用非负矩阵分解等方法来捕捉跨细胞类型的梯度。PME模型的灵活性允许其适应许多类型的细胞状态,包括沿单细胞轨迹的非线性伪时间或通过投影到参考上的状态。



















 799
799

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











