







 本文介绍了scBasset,一种利用CNN处理单细胞ATAC-seq数据的方法,关注DNA序列信息,通过卷积层捕捉TFmotifs和序列特征,预测染色质可及性。模型结构包括1D卷积、卷积塔、全连接层,以及批次校正模块用于考虑批次效应。
本文介绍了scBasset,一种利用CNN处理单细胞ATAC-seq数据的方法,关注DNA序列信息,通过卷积层捕捉TFmotifs和序列特征,预测染色质可及性。模型结构包括1D卷积、卷积塔、全连接层,以及批次校正模块用于考虑批次效应。
目录
0 参考文献
scBasset原论文:scBasset: sequence-based modeling of single-cell ATAC-seq using convolutional neural networks
1 模型特点
对于染色质可及性和scATAC-seq数据以及表观组学的介绍已经在之前的博客中提及,这里不再重复。
许多对于scATAC-seq数据进行分析的方法都只关注peak-by-cell矩阵,即每一行是一个peak(peak指的是某些区域的DNA开放性显著高于其他区域,感觉可以翻译为峰值区域),每一列对应一个细胞的矩阵。大多数方法采用这些标记的peak,将它们表示为基因组上的坐标,而忽略了这些peak对应的具体DNA序列信息。例如SCALE,使用VAE+GMM的方法将peak-by-cell矩阵映射到一个低维空间。
这篇文章提出的scBasset是一种应用DNA序列信息,基于卷积神经网络CNNs来处理scATAC-seq数据的方法。
在这个模型中,第一个卷积层学习转录因子结合位点(TF motifs,特定DNA序列)和其他序列因子。后续卷积层计算这些特征的非线性组合,来得到序列的嵌入结果。最后添加线性层以预测单细胞染色质可及性数据。
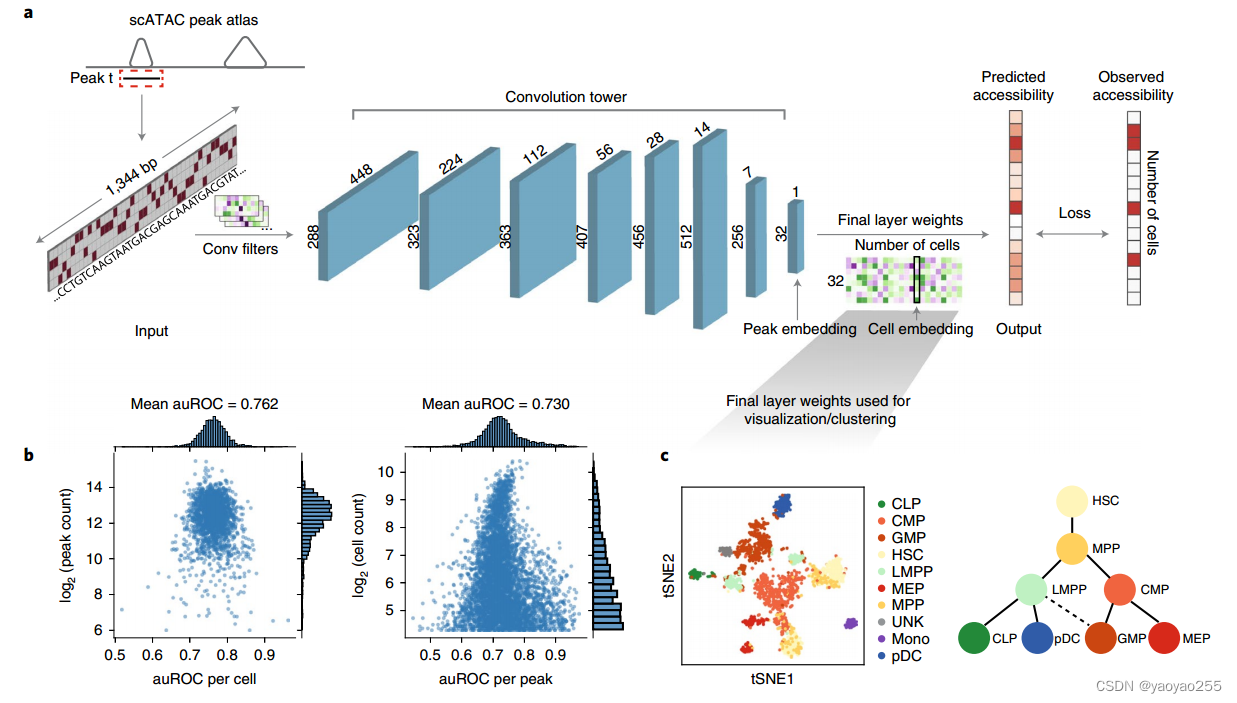
2 模型结构
模型的从每一个peak(很长,具有很多碱基对)的中心选取长度为1344-bp的DNA序列,并使用one-hot转换为一个1344*4的矩阵(因为DNA碱基只有A、T、C、G四种),即每一个peak对应一个1344*4的矩阵。
模型第一层是一个1D卷积层,包含288个大小为17*4的filter(有padding),随后进行批次标准化和GELU激活,再通过宽度为3的最大池化操作,最后输出大小为448*228的矩阵。
随后是一个卷积塔(convolution tower),如上图所示,包含六个卷积单元,每个单元包含卷积层、批次标准化、GELU和最大池化。最终输出矩阵大小为7*512。
接着是一个1D卷积层,包含256个宽度为1的filter,依旧跟随着批次标准化和GELU。输出大小为7*256,并将其展开成1*1792的向量。
将向量输入一个具有32个单元的瓶颈层(批次标准化、0.2的dropout率、GELU),最终输出大小为1*32的向量。这个向量是这个peak(中心1344个碱基序列)的低维表示。
最后通过一个全连接层预测该peak在每个细胞中的开放程度。
该模型具有一个可选模块用于批次校正。在瓶颈层后连接的第二个并行的全连接层,用于预测特定批次的细胞的染色质可及性向量,这个向量会与batch-by-cell矩阵相乘来表示不同批次(例如不同捐献者)对于染色质可及性的影响。这个影响会加到之前预测的开放程度上,并最终以一个sigmoid激活函数放缩到[0,1]范围。
假设有n_peaks个peak,n)cells个细胞。那么瓶颈模块最终得到的表示为n_peaks*32矩阵,通过全连接层学习到的参数则是一个32*n_cells的权重矩阵(忽略截距,因为测序深度信息完全由最终层的截距项所捕获,而不希望考虑测序深度造成的影响),这个矩阵每一列对应一个细胞的低维表示,通过这个矩阵的列向量可以对细胞进行聚类、可视化等操作。
ps:方法上基本就是卷积网络获取低维表示,其实没有太多需要细说的地方。

















 5483
5483

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









