







首先,需要准备两个文件。第一个是建库(近缘物种目标基因),类似于这种格式;第二个是自己物种的protein.fasta

打开TBtools,选择blast→several sequences to a big file
然后,在方框处,上载参考序列,然后在下面选择目标的组学数据,然后填入自己结果的输出路径以及文件命名,开始start
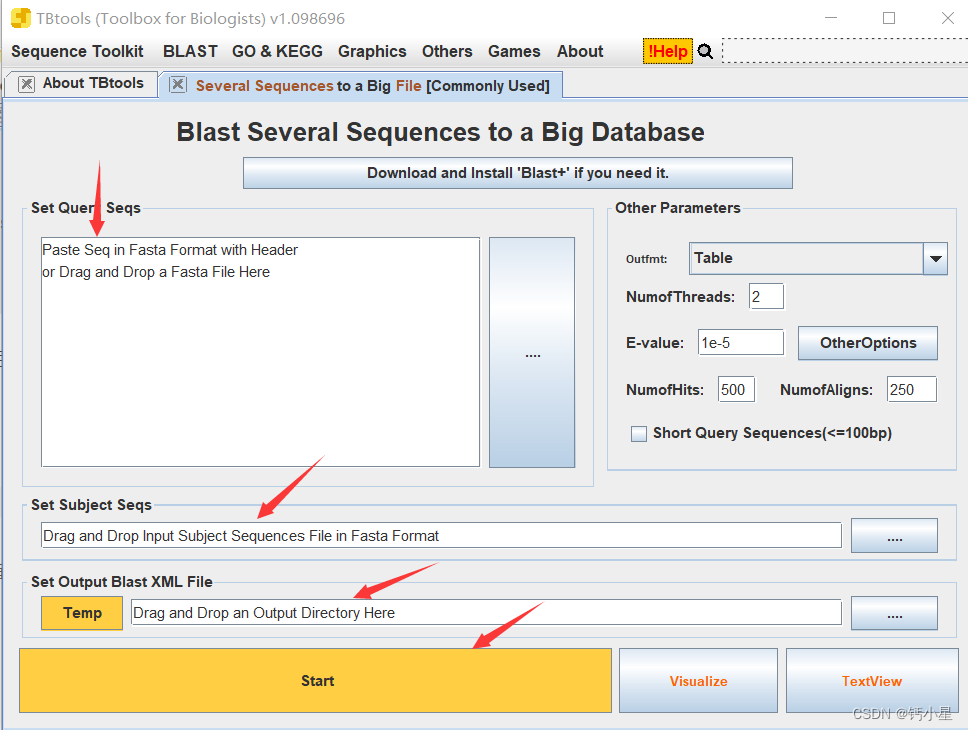
然后得到了结果文件
Query id:查询序列ID标识
Subject id:比对上的目标序列ID标识
% identity:序列比对的一致性百分比
alignment length:符合比对的比对区域的长度
mismatches:比对区域的错配数
gap openings:比对区域的gap数目
q. start:比对区域在查询序列(Query id)上的起始位点
q. end:比对区域在查询序列(Query id)上的终止位点
s. start:比对区域在目标序列(Subject id)上的起始位点
s. end:比对区域在目标序列(Subject id)上的终止位点
e-value:比对结果的期望值
bit score:比对结果的bit score值

















 1751
1751

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









