






上一篇推文介绍了Scanpy流程中的10X数据读取/过滤/降维/聚类步骤,这次笔者将学习一下差异分析/细胞注释/数据保存。
推文链接:https://mp.weixin.qq.com/s/qQ5gq_yvOzu4n1pcL6NMYw
步骤流程
接着上一篇推文内容
1、Clustering the neighborhood graph 构建邻域图
这次创建一个循环函数,可以再IDE中产生多张图片,这样可以挑选自己想要的resolution
# 定义分辨率列表
# 使用 np.arange() 生成从 0.4 到 1.2 之间,步长为 0.2 的分辨率列表
resolutions = np.arange(0.2, 2, 0.2)
# 创建一个字典来保存每个分辨率下的结果
leiden_results = {}
# 循环遍历每个分辨率值
for res in resolutions:
sc.tl.leiden(adata, resolution=res, random_state=0, flavor="igraph", n_iterations=2, directed=False)
# 将结果保存到字典中,键为分辨率值
leiden_results[res] = adata.obs['leiden'].copy()
sc.tl.umap(adata)
sc.pl.umap(adata, color=['leiden'], title=f'Leiden Clustering (resolution={res})')
每一个分辨率都会出一张图,就不展示了
2、检查完上面的图片之后选择想要的resolution值进行umap绘图
# check上面循环的umap图片之后选定自己想要的resolution
sc.tl.leiden(
adata,
resolution=0.8,
random_state=0,
flavor="igraph",
n_iterations=2,
directed=False,
)
3、umap绘图
sc.pl.umap(adata, color=["leiden"])

4、Finding marker genes
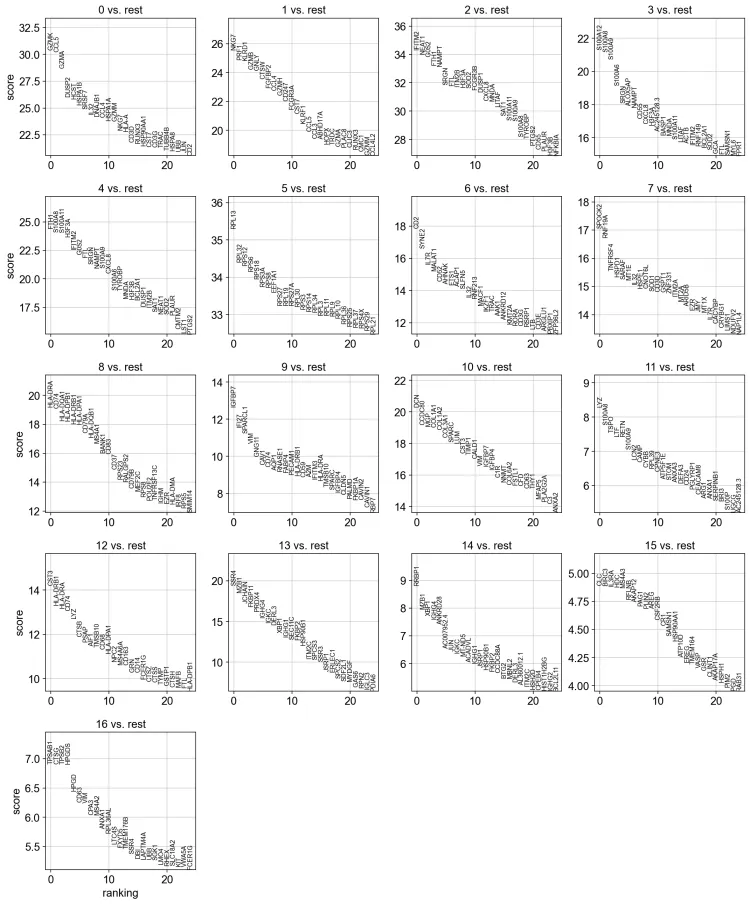
sc.tl.rank_genes_groups(adata, "leiden", method="t-test")
sc.pl.rank_genes_groups(adata, n_genes=25, sharey=False)
sc.tl.rank_genes_groups?
# 在单细胞数据中,根据不同的群体(在这个例子中是使用了 "leiden" 聚类得到的群体)进行差异表达基因的排名。
# "leiden":指定了根据 leiden 聚类标签来区分细胞群体。leiden 是之前运行 sc.tl.leiden() 生成的聚类结果。
# method="t-test":使用 t 检验来确定不同群体之间的差异表达基因。这种方法通过比较不同群体之间的基因表达值,计算每个基因的 p 值,进而排名基因的重要性。
sc.pl.rank_genes_groups?
# 可视化前一步计算得到的差异表达基因排名结果。
# n_genes=25:指定要显示的前 25 个差异表达基因。
# sharey=False:表示在绘制多个基因的表达量分布图时,每个基因的 y 轴不共享,这样可以分别展示每个基因在不同群体中的表达差异。
5、定义marker基因
marker_genes = [
*["IL7R", "CD79A", "MS4A1", "CD8A", "CD8B", "LYZ", "CD14"],
*["LGALS3", "S100A8", "GNLY", "NKG7", "KLRB1"],
*["FCGR3A", "MS4A7", "FCER1A", "CST3", "PPBP"],
]
6、提取每一簇中的关键基因
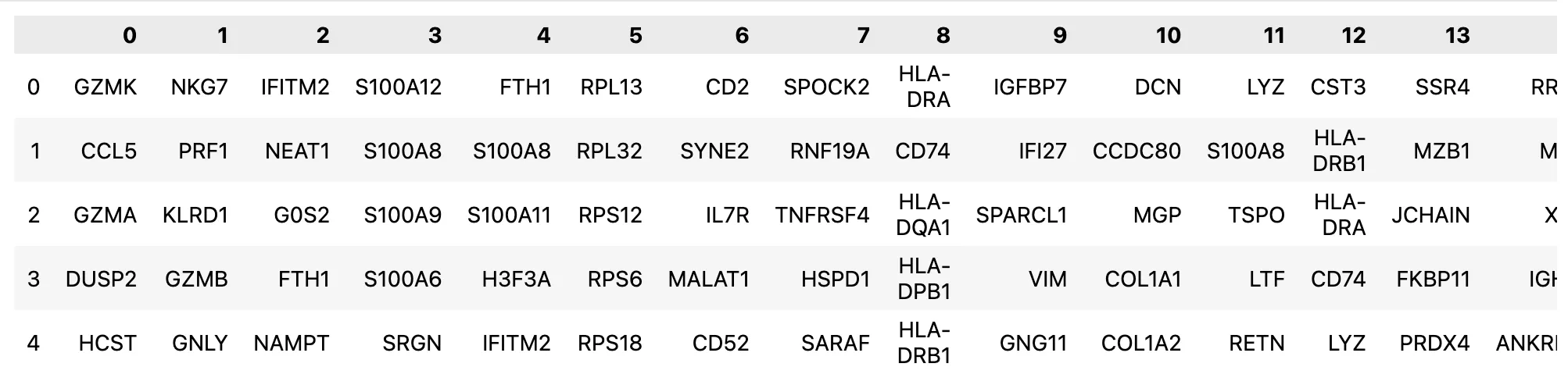
pd.DataFrame(adata.uns["rank_genes_groups"]["names"]).head(5)
# adata.uns["rank_genes_groups"]["names"]是什么意思
# uns 是 AnnData 对象的一个属性,表示 “unstructured annotations”(非结构化注释)。通常用于存储一些不属于 obs、var 或 obsm 等的注释或计算结果。
# "rank_genes_groups" 是在 uns 中的一个键(key),通常用于存储通过 sc.tl.rank_genes_groups 计算的差异表达基因的结果。
# "names" 是在 rank_genes_groups 中的一个键,通常用于存储排名靠前的基因名(即在各个分组中最显著的基因)。
# pd 是 pandas 库的别名,DataFrame 是 pandas 中的一个类,用于表示二维的表格数据结构。
# pd.DataFrame(adata.uns["rank_genes_groups"]["names"]) 将 adata.uns["rank_genes_groups"]["names"] 中的数据转换为一个 DataFrame 对象,方便数据操作和分析。
# head() 是 pandas DataFrame 的一个方法,用于返回前 n 行数据,默认值是 5。head(5) 表示返回 DataFrame 的前五行。
7、获得每个簇中基因的名称和p值
result = adata.uns["rank_genes_groups"]
groups = result["names"].dtype.names
pd.DataFrame(
{
group + "_" + key[:1]: result[key][group]
for group in groups
for key in ["names", "pvals"]
}
).head(5)
# result 是一个字典数据结构,它包含了分析的结果。
# ["names"] 是从 result 中获取名为 "names" 的数据。这个数据是一个结构化数组,记录了多个组的基因名称。
# dtype 是数据类型对象,它描述了 NumPy 数组的元素类型。
# 1. key的定义:
# key 是在 for key in ["names", "pvals"] 这部分循环中定义的。
# 这个循环会依次将 key 设置为 "names" 和 "pvals"。
# 2. result[key][group] 的作用:
# result 是 adata.uns["rank_genes_groups"],这个数据结构通常用于存储差异表达基因分析的结果,包含多个分组(group)的信息。
# result["names"] 和 result["pvals"] 分别存储了与分组相关的基因名称(names)和它们的 p 值(pvals)。
# 3. key[:1]:
# key[:1] 取的是 key 的第一个字符。
# 当 key 是 "names" 时,key[:1] 是 "n"。
# 当 key 是 "pvals" 时,key[:1] 是 "p"。
# 4. 具体操作:
# 对于每个 group(例如 "group1"),这段代码会生成两个键值对:
# 一个键是 "group1_n",对应的值是 result["names"]["group1"]。
# 另一个键是 "group1_p",对应的值是 result["pvals"]["group1"]。
# 这个过程会对所有的 group 和 key 进行循环,从而创建出一个完整的字典,最终将这个字典转换成一个 pandas DataFrame。

8、单个簇比较
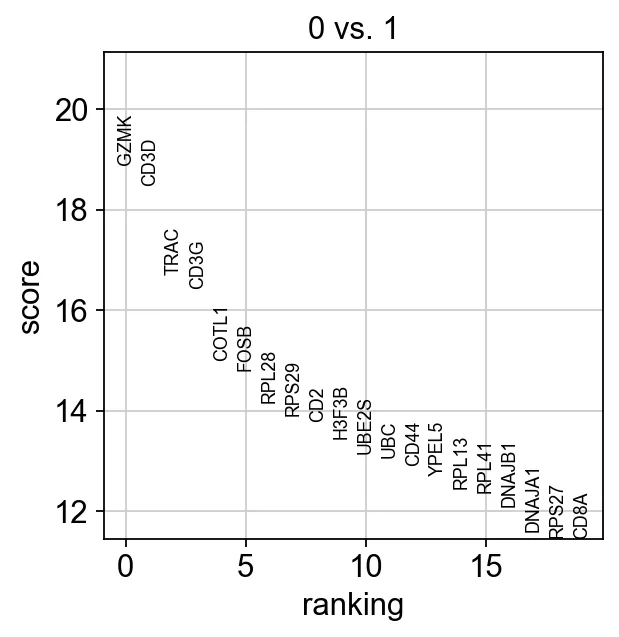
sc.tl.rank_genes_groups(adata, "leiden", groups=["0"], reference="1", method="wilcoxon")
sc.pl.rank_genes_groups(adata, groups=["0"], n_genes=20)
9、绘制一下两组的小提琴图
sc.pl.rank_genes_groups_violin(adata, groups="0", n_genes=8)
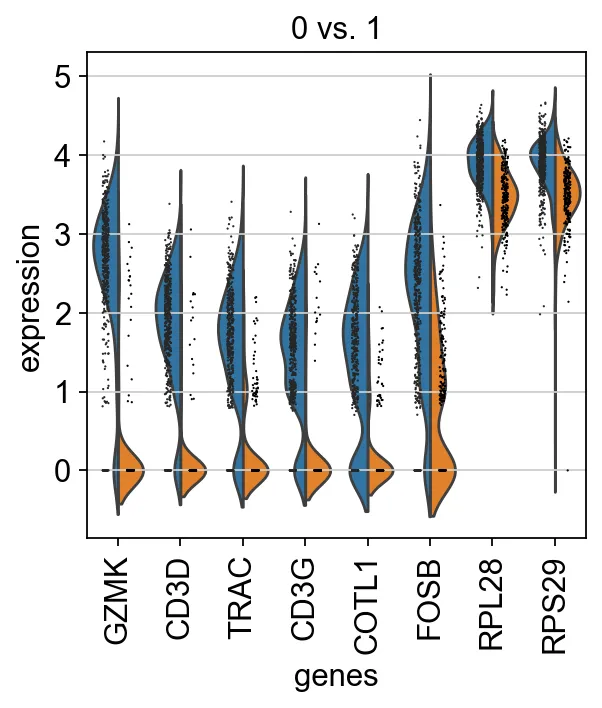
10、绘制一下不同基因在不同簇中的小提琴图
sc.pl.violin(adata, ["CST3", "NKG7", "PPBP"], groupby="leiden")
11、重命名
请注意笔者这里没有按照数量和正确的marker去命名,后面的分簇情况没有参考价值。
笔者尝试了三种方式,也解决了三种报错。
1、命名数要与分簇数一致,不可多或者少。
2、命名不可重复。
new_cluster_names = [
"CD4 T",
"B",
"FCGR3A+ Monocytes",
"NK",
"CD8 T",
"CD14+ Monocytes",
"Dendritic",
"Megakaryocytes",
"CD4 T1",
"B1",
"FCGR3A1+ Monocytes",
"NK1",
"CD81 T",
"CD141+ Monocytes",
"Dendritic1",
"Megakaryocytes1",
"Megakaryocytes2"
]
adata.rename_categories("leiden", new_cluster_names)
sc.pl.umap(
adata, color="leiden", legend_loc="on data", title="", frameon=False, save=".pdf"
)
是胡乱命名的。但是这样有一个问题,假设好几个簇可能是同一种细胞怎么办呢?

3、分群数过多,多个簇分成同一种细胞,通过映射的方式
# 定义映射,将多个簇合并为一个
cluster_mapping = {
'0': 'CD4 T',
'1': 'CD4 T',
'2': 'CD4 T',
'3': 'NK',
'4': 'CD8 T',
'5': 'CD14+ Monocytes',
'6': 'Dendritic',
'7': 'Megakaryocytes',
'8': 'CD4 T',
'9': 'CD4 T',
'10': 'NK',
'11': 'CD8 T',
'12': 'CD14+ Monocytes',
'13': 'Dendritic',
'14': 'Megakaryocytes',
'15': 'Dendritic',
'16': 'Megakaryocytes',
# 继续映射其他簇
}
# 应用映射
adata.obs['leiden1'] = adata.obs['leiden'].map(cluster_mapping)
sc.pl.umap(
adata, color="leiden1", legend_loc="on data", title="", frameon=False, save=".pdf"
)
命名成功

12、dotplot
sc.pl.dotplot(adata, marker_genes, groupby="leiden");
请注意,笔者这边是随便注释的哈!

13、绘制小提琴图
sc.pl.stacked_violin(adata, marker_genes, groupby="leiden");
请注意,笔者这边是随便注释的哈!
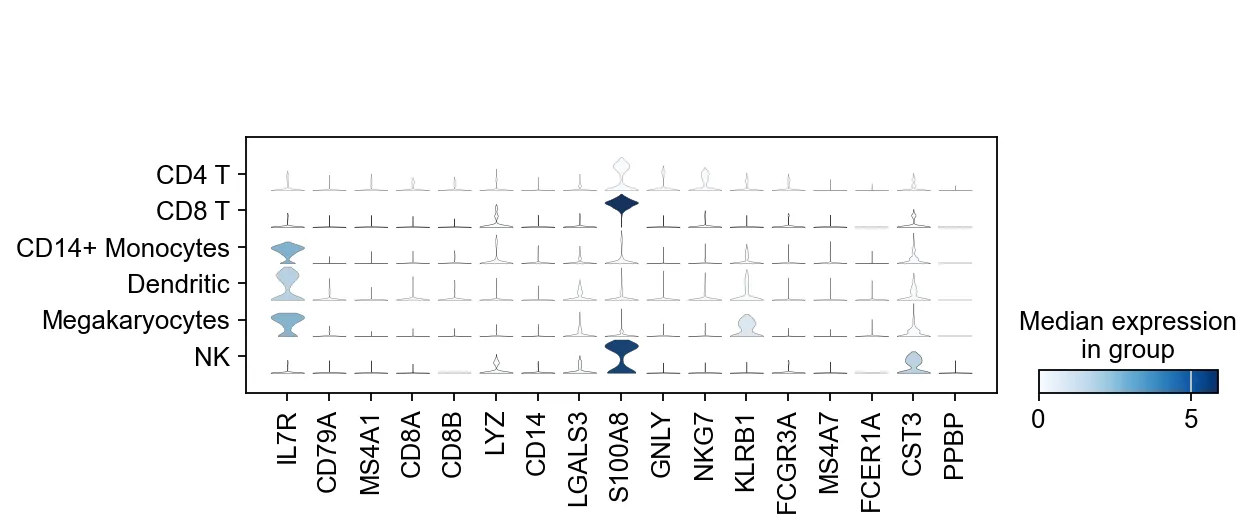
14、创建文件夹+保存h5ad文件
请注意这种保存方式不会保存降维聚类之后的数据结果
os.makedirs("output",exist_ok = True)
adata.raw.to_adata().write("./output/test.h5ad")
这种方式是保存了处理之后的数据结果
adata.write("./output/test_none.h5ad")

整理一下代码
# 加载库
import pandas as pd
import scanpy as sc
import numpy as np
import anndata as ad
import pooch
# 参数设置
sc.settings.verbosity = 3
sc.logging.print_header()
sc.settings.set_figure_params(dpi=80, facecolor="white")
# 读取数据
adata = sc.read_10x_mtx(
"input/",
var_names="gene_symbols",
cache=True,
)
# 让基因名称不重复
adata.var_names_make_unique()
# 基础过滤
sc.pp.filter_cells(adata, min_genes=200)
sc.pp.filter_genes(adata, min_cells=3)
# 增加线粒体基因数据
adata.var["mt"] = adata.var_names.str.startswith("MT-")
sc.pp.calculate_qc_metrics(
adata, qc_vars=["mt"], percent_top=None, log1p=False, inplace=True
)
# 基础数据绘图
sc.pl.violin(
adata,
["n_genes_by_counts", "total_counts", "pct_counts_mt"],
jitter=0.4,
multi_panel=True,
)
# 基础数据绘图
sc.pl.scatter(adata, x="total_counts", y="pct_counts_mt")
sc.pl.scatter(adata, x="total_counts", y="n_genes_by_counts")
# 过滤
adata = adata[adata.obs.n_genes_by_counts < 2500, :]
adata = adata[adata.obs.pct_counts_mt < 5, :].copy()
# 根据上述信息可以调整过滤参数
##################################
# Normilize+log数据
sc.pp.normalize_total(adata, target_sum=1e4)
sc.pp.log1p(adata)
# 寻找高变基因
sc.pp.highly_variable_genes(adata, min_mean=0.0125, max_mean=3, min_disp=0.5)
# 高变基因绘图
sc.pl.highly_variable_genes(adata)
# 备份一下
adata.raw = adata
# scale数据
sc.pp.scale(adata, max_value=10)
# pca降维
sc.tl.pca(adata, svd_solver="arpack")
# 计算邻域图+umap处理
sc.pp.neighbors(adata, n_neighbors=10, n_pcs=40)
sc.tl.umap(adata)
#################
# 定义分辨率列表
# 使用 np.arange() 生成从 0.4 到 1.2 之间,步长为 0.2 的分辨率列表
resolutions = np.arange(0.2, 2, 0.2)
# 创建一个字典来保存每个分辨率下的结果
leiden_results = {}
# 循环遍历每个分辨率值
for res in resolutions:
sc.tl.leiden(adata, resolution=res, random_state=0, flavor="igraph", n_iterations=2, directed=False)
# 将结果保存到字典中,键为分辨率值
leiden_results[res] = adata.obs['leiden'].copy()
sc.tl.umap(adata)
sc.pl.umap(adata, color=['leiden'], title=f'Leiden Clustering (resolution={res})')
# check上面循环的umap图片之后选定自己想要的resolution
sc.tl.leiden(
adata,
resolution=0.8,
random_state=0,
flavor="igraph",
n_iterations=2,
directed=False,
)
# check图形
sc.pl.umap(adata, color=["leiden"])
# 找marker基因
sc.tl.rank_genes_groups(adata, "leiden", method="wilcoxon")
sc.pl.rank_genes_groups(adata, n_genes=25, sharey=False)
# 定义marker基因
marker_genes = [
"IL7R", "CD79A", "MS4A1", "CD8A", "CD8B", "LYZ", "CD14",
"LGALS3", "S100A8", "GNLY", "NKG7", "KLRB1",
"FCGR3A", "MS4A7", "FCER1A", "CST3", "PPBP"],
]
# check一下
pd.DataFrame(adata.uns["rank_genes_groups"]["names"]).head(5)
# check两下
result = adata.uns["rank_genes_groups"]
groups = result["names"].dtype.names
pd.DataFrame(
{
group + "_" + key[:1]: result[key][group]
for group in groups
for key in ["names", "pvals"]
}
).head(5)
# 簇与簇之间的基因比较
sc.tl.rank_genes_groups(adata, "leiden", groups=["0"], reference="1", method="wilcoxon")
sc.pl.rank_genes_groups(adata, groups=["0"], n_genes=20
# 簇间差异基因小提琴图展示
sc.pl.rank_genes_groups_violin(adata, groups="0", n_genes=8)
# 小提琴图绘制在不同簇中的基因表达情况
sc.pl.violin(adata, ["CST3", "NKG7", "PPBP"], groupby="leiden")
# 细胞注释
# 定义映射,将多个簇合并为一个
cluster_mapping = {
'0': 'CD4 T',
'1': 'CD4 T',
'2': 'CD4 T',
'3': 'NK',
'4': 'CD8 T',
'5': 'CD14+ Monocytes',
'6': 'Dendritic',
'7': 'Megakaryocytes',
'8': 'CD4 T',
'9': 'CD4 T',
'10': 'NK',
'11': 'CD8 T',
'12': 'CD14+ Monocytes',
'13': 'Dendritic',
'14': 'Megakaryocytes',
'15': 'Dendritic',
'16': 'Megakaryocytes',
# 继续映射其他簇
}
# 应用映射
adata.obs['leiden1'] = adata.obs['leiden'].map(cluster_mapping)
# 注释后绘图
sc.pl.umap(
adata, color="leiden1", legend_loc="on data", title="", frameon=False, save=".pdf"
)
# 创建输出文件+保存数据
os.makedirs("output",exist_ok = True)
adata.write("./output/test_none.h5ad")
注:若对内容有疑惑或者有发现明确错误的朋友,请联系后台(欢迎交流)。更多内容可关注公众号:生信方舟
- END -

















 376
376

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









