







ngs-bits - Short-read sequencing tools - 短reasd测序工具
一个强大的工具集,安装后直接使用。
# conda安装指定版本, 安装高版本失败,故采用2021_09版本
conda install ngs-bits=2021_09 -y
# 测试安装是否成功,成功则显示下列内容
ReadQC
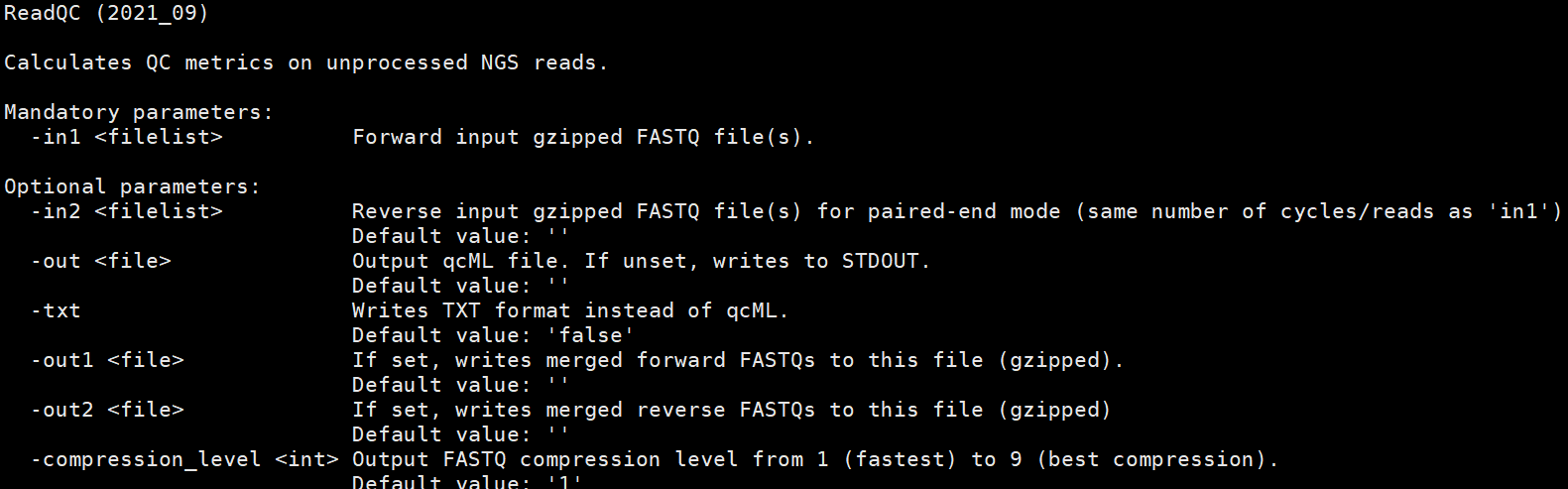
软件说明文档markown
# 参考github
https://github.com/imgag/ngs-bits
SampleGender - 样本性别判断实例
Determines the gender of a sample from the BAM/CRAM file.
Mandatory parameters:
-in <filelist> Input BAM/CRAM file(s).
-method <enum> Method selection: Read distribution on X and Y chromosome (xy), fraction of heterozygous variants on X chromosome (hetx), or coverage of SRY gene (sry).
Valid: 'xy,hetx,sry'
Optional parameters:
-out <file> Output TSV file - one line per input BAM/CRAM file. If unset, writes to STDOUT.
Default value: ''
-max_female <float> Maximum Y/X ratio for female (method xy).
Default value: '0.06'
-min_male <float> Minimum Y/X ratio for male (method xy).
Default value: '0.09'
-min_female <float> Minimum heterozygous SNP fraction for female (method hetx).
Default value: '0.25'
-max_male <float> Maximum heterozygous SNP fraction for male (method hetx).
Default value: '0.05'
-sry_cov <float> Minimum average coverage of SRY gene for males (method sry).
Default value: '20'
-build <enum> Genome build used to generate the input (methods hetx and sry).
Default value: 'hg38'
Valid: 'hg19,hg38'
-ref <file> Reference genome for CRAM support (mandatory if CRAM is used).
Default value: ''
Special parameters:
--help Shows this help and exits.
--version Prints version and exits.
--changelog Prints changeloge and exits.
--tdx Writes a Tool Definition Xml file. The file name is the application name with the suffix '.tdx'.
使用方法
# 创建idx
samtools index sample.sorted.bam
# 基于xy染色体 reads数量
SampleGender -in sample.sorted.bam -method xy
# female

# 基于x染色体上杂合位点的比例
SampleGender -in sample.sorted.bam -method hetx

# 基于Y染色体sry基因的覆盖度
SampleGender -in sample.sorted.bam -method sry

其他工具介绍
主要工具
SeqPurge - A highly-sensitive adapter trimmer for paired-end short-read data.
Seqtek-一种用于双端短读数据的高灵敏度适配器微调器。
SampleSimilarity - Calculates pairwise sample similarity metrics from VCF/BAM files.
SampleSimilarity-计算VCF/BAM文件中的成对样本相似性度量。
SampleGender - Determines sample gender based on a BAM file.
SampleGender-根据BAM文件确定样本性别。
SampleAncestry - Estimates the ancestry of a sample based on variants.
Samplestry-基于变量估计样本的祖先。
CnvHunter - CNV detection from targeted resequencing data using non-matched control samples.
CnvHunter-使用非匹配对照样品从靶向重测序数据中检测CNV。
RohHunter - ROH detection based on a variant list annotated with AF values.
RohHunter-基于标注AF值的变体列表的ROH检测。
UpdHunter - UPD detection from trio variant data.
UpdHunter-从三个变体数据中检测UPD。
质量控制工具
ReadQC - Quality control tool for FASTQ files.
ReadQC-FASTQ文件的质量控制工具。
MappingQC - Quality control tool for a BAM file.
MappingQC-BAM文件的质量控制工具。
VariantQC - Quality control tool for a VCF file.
VariantQC-VCF文件的质量控制工具。
SomaticQC - Quality control tool for tumor-normal pairs
( 肿瘤-正常配对的质量控制工具)
TrioMaternalContamination - Detects maternal contamination of a child using SNPs from parents.
TrioMaternalContamination-使用父母的SNP检测儿童的母体污染。
RnaQC - Calculates QC metrics for RNA samples.
RnaQC-计算RNA样本的QC指标。
BAM工具
BamClipOverlap - (Soft-)Clips paired-end reads that overlap.
BamClipOverlap-(Soft-)剪辑重叠的双端读取。
BamDownsample - Downsamples a BAM file to the given percentage of reads.
BamDownsample-将BAM文件下采样到给定的读取百分比。
BamFilter - Filters a BAM file by multiple criteria.
BamFilter-按多个条件过滤BAM文件。
BamHighCoverage - Determines high-coverage regions in a BAM file.
BamHighCoverage-确定BAM文件中的高覆盖率区域。
BamToFastq - Converts a BAM file to FASTQ files (paired-end only).
BamToFastq-将BAM文件转换为FASTQ文件(仅适用于双端)。
BED工具
BedAdd - Merges regions from several BED files.
BedAdd-合并多个BED文件中的区域。
BedAnnotateFromBed - Annotates BED file regions with information from a second BED file.
BedAnnotateFromBed-使用来自第二个BED文件的信息注释BED文件区域。
BedAnnotateGC - Annnotates the regions in a BED file with GC content.
BedAnnotateGC-用GC内容在BED文件中Annotates区域。
BedAnnotateGenes - Annotates BED file regions with gene names (needs - 用基因名称注释BED文件区域(需要 NGSD)
BedChunk - Splits regions in a BED file to chunks of a desired size.
BedChunk-将BED文件中的区域拆分为所需大小的块。
BedCoverage - Annotates the regions in a BED file with the average coverage in one or several BAM files.
BedCoverage-使用一个或多个BAM文件中的平均覆盖率注释BED文件中的区域。
BedExtend - Extends the regions in a BED file by n bases.
BedExtend-将BED文件中的区域扩展n个碱基。
BedGeneOverlapBedGeneOverlap的 - Calculates how much of each overlapping gene is covered (needs - 计算每个重叠基因的覆盖量(需要 NGSD).
BedHighCoverage - Detects high-coverage regions from a BAM file.
BedHighCoverage-从BAM文件中检测高覆盖率区域。
BedInfo - Prints summary information about a BED file.
BedInfo-打印有关BED文件的摘要信息。
BedIntersect - Intersects two BED files.
BedIntersect-使两个BED文件相交。
BedLiftOver - Lift-over of regions in a BED file to a different genome build.
BedLiftOver-将BED文件中的区域转移到不同的基因组构建。
BedLowCoverage - Calcualtes regions of low coverage based on a input BED and BAM file.
BedLowCoverage-基于输入BED和BAM文件计算低覆盖率区域。
BedMerge - Merges overlapping regions in a BED file.
BedMerge-合并BED文件中的重叠区域。
BedReadCount - Annoates the regions in a BED file with the read count from a BAM file.
BedReadCount-使用BAM文件中的读取计数注释BED文件中的区域。
BedShrink - Shrinks the regions in a BED file by n bases.
BedShrink-将BED文件中的区域收缩n个碱基。
BedSort - Sorts the regions in a BED file
BedSort-对BED文件中的区域进行排序
BedSubtract - Subracts one BED file from another BED file.
从一个BED文件中减去另一个BED文件。
BedToFasta - Converts BED file to a FASTA file (based on the reference genome).
BedToFasta-将BED文件转换为FASTA文件(基于参考基因组)。
FASTQ工具
FastqAddBarcode - Adds sequences from separate FASTQ as barcodes to read IDs.
FastqAddBarcode-将来自单独FASTQ的序列作为条形码添加到读取ID。
FastqConvert - Converts the quality scores from Illumina 1.5 offset to Sanger/Illumina 1.8 offset.
FastqConvert-将质量分数从Illumina 1.5偏移调整为桑格/Illumina 1.8偏移。
FastqConcat - Concatinates several FASTQ files into one output FASTQ file.
FastqConcat-将多个FASTQ文件合并为一个输出FASTQ文件。
FastqDownsample - Downsamples paired-end FASTQ files.
FastqDownsample-对双端FASTQ文件进行下采样。
FastqExtract - Extracts reads from a FASTQ file according to an ID list.
FastqExtract-根据ID列表从FASTQ文件中提取读取。
FastqExtractBarcode - Moves molecular barcodes of reads to a separate file.
FastqExtractBarcode-将读取的分子条形码移动到单独的文件中。
FastqExtractUMI - Moves unique moleculare identifier from read sequence to read ID.
FastqExtractUMI-将唯一分子标识符从读取序列移动到读取ID。
FastqFormat - Determines the quality score offset of a FASTQ file.
FastqFormat-确定FASTQ文件的质量分数偏移。
FastqList - Lists read IDs and base counts.
FastqList-列出读取ID和碱基计数。
FastqMidParser - Counts the number of occurances of each MID/index/barcode in a FASTQ file.
FastqMidParser-计算FASTQ文件中每个MID/索引/条形码的出现次数。
FastqToFasta - Converts FASTQ to FASTA format.
FastqToFasta-将FASTQ转换为FASTA格式。
FastqTrim - Trims start/end bases from the reads in a FASTQ file.
FastqTrim-从FASTQ文件中的读段修剪起始/结束碱基。
VCF工具
VcfAdd - Appends variants from a VCF file to another VCF file.
VcfAdd-将变量从一个VCF文件转换到另一个VCF文件。
VcfAnnotateConsequence - Adds transcript-specific consequence predictions to a VCF file (similar to Ensembl VEP).
VcfAnnotateConsequence-将转录本特定的结果预测添加到VCF文件(类似于Ensembl VEP)。
VcfAnnotateFromBed - Annotates the INFO column of a VCF with data from a BED file.
VcfAnnotateFromBed-使用来自BED文件的数据注释VCF的INFO列。
VcfAnnotateFromBigWig - Annotates the INFO column of a VCF with data from a BED file.
VcfAnnotateFromBigWig-使用来自BED文件的数据注释VCF的INFO列。
VcfAnnotateFromVcf - Annotates a VCF file with data from one or more source VCF files.
VcfAnnotateFromVcf-使用来自一个或多个源VCF文件的数据注释VCF文件。
VcfAnnotateHexplorer - Annotates a VCF with Hexplorer and HBond scores.
VcfAnnotateHexplorer-使用Hexplorer和HBOND分数注释VCF。
VcfAnnotateMaxEntScan - Annotates a VCF file with MaxEntScan scores.
VcfAnnotateMaxEntScan-使用MaxEntScan分数注释VCF文件。
VcfBreakMulti - Breaks multi-allelic variants into several lines, making sure that allele-specific INFO/SAMPLE fields are still valid.
VcfBreakMulti-将多等位基因变体分成几行,确保等位基因特定的INFO/SAMPLE字段仍然有效。
VcfCalculatePRS - Calculates the Polgenic Risk Score(s) for a sample.
VcfCalculatePRS-计算样本的Polgenic风险评分。
VcfCheck - Checks a VCF file for errors.
VcfCheck-检查VCF文件的错误。
VcfExtractSamples - Extract one or several samples from a VCF file.
VcfExtractSamples-从VCF文件中提取一个或多个样本。
VcfFilter - Filters a VCF based on the given criteria.
VcfFilter-根据给定条件过滤VCF。
VcfLeftNormalize - Normalizes all variants and shifts indels to the left in a VCF file.
VcfLeftNormalize-规范化所有变量,并将VCF文件中的indel向左移动。
VcfSort - Sorts variant lists according to chromosomal position.
VcfSort-根据染色体位置对变体列表进行排序。
VcfStreamSort - Sorts entries of a VCF file according to genomic position using a stream.
VcfStreamSort-使用流根据基因组位置对VCF文件的条目进行排序。
VcfSubstract - Substracts the variants in a VCF from a second VCF.
从第二个VCF中减去VCF中的变量。
VcfToBed - Converts a VCF file to a BED file.
VcfToBed-将VCF文件转换为BED文件。
VcfToBedpe - Converts a VCF file containing structural variants to BEDPE format.
VcfToBedpe-将包含结构变体的VCF文件转换为BEDPE格式。
VcfToTsv - Converts a VCF file to a tab-separated text file.
将VCF文件转换为制表符分隔的文本文件。
SV工具(结构变异工具)
BedpeAnnotateFromBed - Annotates a BEDPE file with information from a BED file.
BedpeAnnotateFromBed-使用来自BED文件的信息注释BEDPE文件。
BedpeFilter - Filters a BEDPE file by region.
BedpeFilter-按区域过滤BEDPE文件。
BedpeGeneAnnotation - Annotates a BEDPE file with gene information from the NGSD (needs - 用NGSD中的基因信息注释BEDPE文件(需要 NGSD).).
BedpeSort - Sort a BEDPE file according to chromosomal position.
BedpeSort-根据染色体位置对BEDPE文件进行排序。
BedpeToBed - Converts a BEDPE file into BED file.
BedpeToBed-将BEDPE文件转换为BED文件。
SvFilterAnnotations - Filter a structural variant list in BEDPE format based on variant annotations.
SvFilterAnnotations-基于变量注释过滤BEDPE格式的结构变量列表。
基因处理工具
GenePrioritization: Performs gene prioritization based on list of known disease genes and a PPI graph (see also GraphStringDb).
GenePrioritization:基于已知疾病基因列表和PPI图执行基因优先级排序(参见GraphStringDb)。
GraphStringDb: Creates simple representation of String-DB interaction graph.
GraphStringDb:创建String-DB交互图的简单表示。
GenesToApproved - Replaces gene symbols by approved symbols using the HGNC database
(needs - 使用HGNC数据库将基因符号替换为批准的符号(需要 NGSD).
GenesToBed创世记 - Converts a text file with gene names to a BED file (needs - 将包含基因名称的文本文件复制到BED文件(需要 NGSD).)
GenesToTranscripts基因转转录 - Converts a text file with gene names to transcript names (needs - 将带有基因名称的文本文件转换为转录名称(需要 NGSD).)
NGSDExportGenesNGSD出口基因 - Lists genes from NGSD (needs - 列出NGSD中的基因(需要 NGSD).).
TranscriptsToBed - Converts a text file with transcript names to a BED file (needs - 将带有成绩单名称的文本文件复制到BED文件(需要 NGSD).).
表型处理工具
PhenotypesToGenes表型到基因 - Converts a phenotype list to a list of matching genes (needs - 将表型列表转换为匹配基因列表(需要 NGSD).).
PhenotypeSubtree表型子树 - Returns all sub-phenotype of a given phenotype (needs - 返回给定表型的所有子表型(需要 NGSD).).
生信软件文章推荐
生信软件1 - 测序下机文件比对结果可视化工具 visNano
生信软件3 - mapping比对bam文件质量评估工具 qualimap
生信软件4 - 拷贝数变异CNV分析软件 WisecondorX
生信软件7 - 多线程并行运行Linux效率工具Parallel
生信软件8 - bedtools进行窗口划分、窗口GC含量、窗口测序深度和窗口SNP统计
生信软件9 - 多公共数据库数据下载软件Kingfisher
生信软件10 - DNA/RNA/蛋白多序列比对图R包ggmsa
生信软件11 - 基于ACMG的CNV注释工具ClassifyCNV
生信软件12 - 基于Symbol和ENTREZID查询基因注释的R包(easyConvert )
生信软件13 - 基于sambamba 窗口reads计数和平均覆盖度统计
生信软件14 - bcftools提取和注释VCF文件关键信息



















 2841
2841

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











