







1. mrbait安装
mrbait支持linux和macOS系统,测试安装Python版本 3.6.15。
# conda安装
conda install mrbait -c tylerkchafin -c bioconda -c conda-forge
# github安装
git clone https://github.com/tkchafin/mrbait.git
cd mrbait
python ./setup.py install
# 安装依赖库
pip install PyVCF
pip install pandas
pip install biopython
pip install networkx
# 测试安装是否成功
mrbait -h
2. 工作流
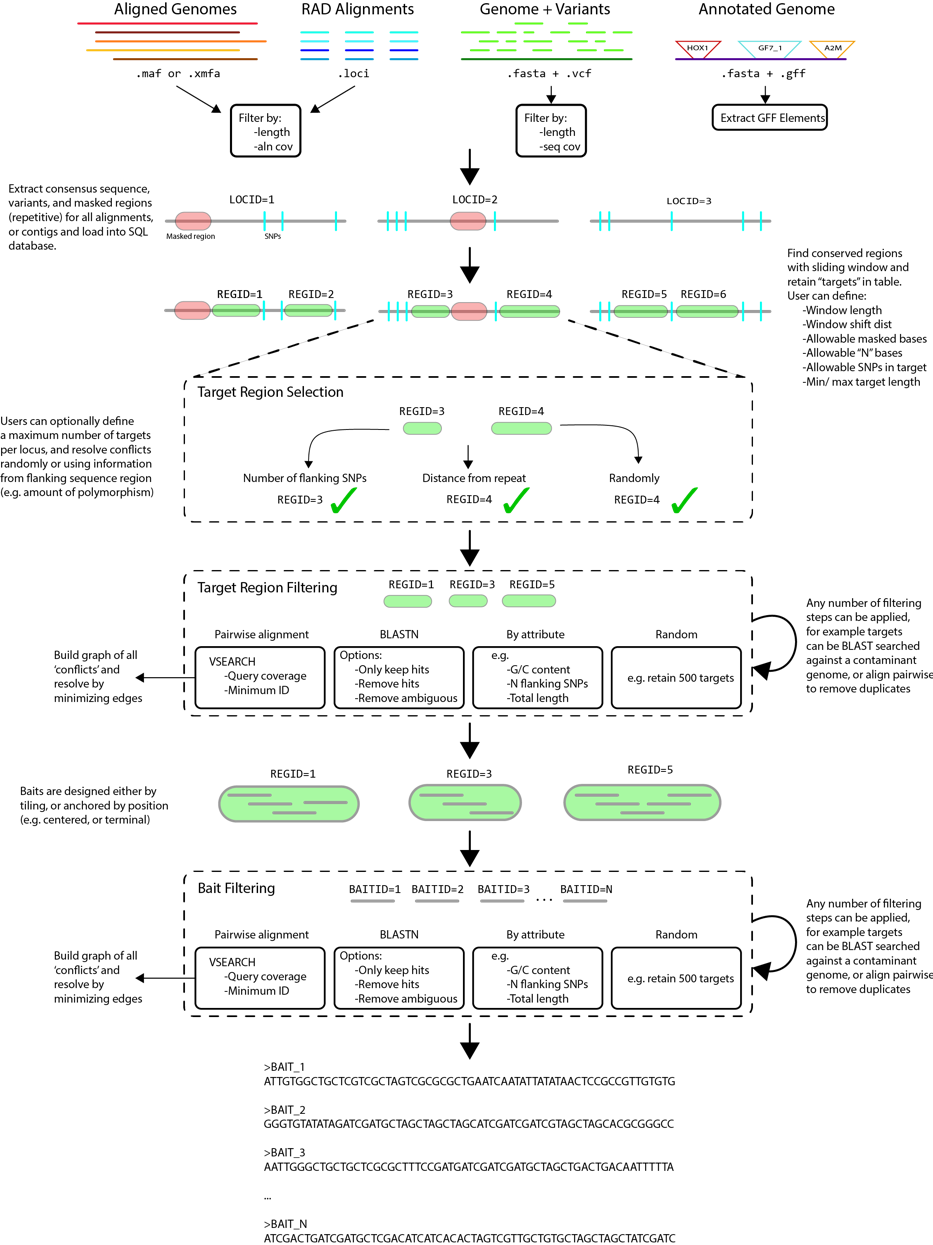
mrbait工作流分为以下5个步骤:
- Alignments (provided as .xmfa, .loci, or .maf) or genomes (provided as .fasta, annotated with .vcf or .gff) will be used to build a consensus sequence of each locus.
- A sliding window will be applied to each consensus to find candidate targets for which baits could be designed
- Targets are then selected (if too close together, or only one allowed per locus), and filtered according to any number of specified filter (e.g. GC content, flanking SNPs, pairwise alignment)
- Passing targets are then parsed to design a putative set of baits
Baits are then filtered according to selected criteria, and output as FASTA. - The pipeline can be resumed and any steps iteratively re-visited by providing the SQLite database file (resulting in a significant reduction in runtime for successive runs)
3.基本用法
mrbait适用于多种输入格式,有maf,vcf,gff,fasta等,通常会选择输入fasta文件进行探针设计。
根据input.fa文件,生成探针长度为120,跨靶区域平铺,重叠40个碱基, 输出文件前缀probe.design
mrbait -A input.fa -b 120 -o probe.design -T 4 -s tile=40
 部分结果:
部分结果:
>HBA_chr16_222006_224006:0-20011:0-120_Bait=1
GTTCAACCAGGGAGATTTCAAAGGAGGGTGGCGATGCTGGGACTCCCTGAGTAGAACCCTGGATGCCTCTCTCCTCCCCAGCTTCTGAGCCACTGCCTGCAGGCCTGGCACCTCTCAGGA
>HBA_chr16_222006_224006:0-20011:80-200_Bait=2
GCTTCTGAGCCACTGCCTGCAGGCCTGGCACCTCTCAGGACAGGGGATGGTTCAGCACCTTCTCTTGAGCAGGCCCACCTCAGCTTCCCCTCCCATACTCCCTGCAGTTCTCCCTCCCCA
>HBA_chr16_222006_224006:0-20011:160-280_Bait=3
CAGCTTCCCCTCCCATACTCCCTGCAGTTCTCCCTCCCCAGCCCCATGCAGCTGCAGAGAGGTCCTTGGTCTGAGACAGGTAAACACCTCCATTGTTGGCACATTCCGGGATAGAGAGAA
......
根据example.maf文件,生成探针长度为80,跨靶区域平铺,重叠40个碱基
mrbait -M example.maf -b 80 -s tile=40
4. 输出文件
mrbait输出文件为格式化的FASTA文件,命名为 o u t b a i t s . f a s t a (其中 out_baits.fasta (其中 outbaits.fasta(其中out使用-o/-out参数定义)。
默认情况
默认情况下,输出全部模糊性序列(例如,作为 consensus sequence):
>Locus1_Target4_Bait1
ATGTAATRAGGTATATG……
>Locus1_Target4_Bait2
TATGAATGTCGCGCGAT……
…
所有组合
如果使用-x/-expand选项,输出所有组合,如下所示:
>Locus2_Target4_Bait1.1
ATGTAATAAGGTATATG……
>Locus2_Target4_Bait1.1
ATGTAATGAGGTATATG……
>Locus1_Target4_Bait2.1
TATGAATGTCGCGCGAT……
…
生信软件文章推荐
生信软件1 - 测序下机文件比对结果可视化工具 visNano
生信软件3 - mapping比对bam文件质量评估工具 qualimap
生信软件4 - 拷贝数变异CNV分析软件 WisecondorX
生信软件7 - 多线程并行运行Linux效率工具Parallel
生信软件8 - bedtools进行窗口划分、窗口GC含量、窗口测序深度和窗口SNP统计
生信软件9 - 多公共数据库数据下载软件Kingfisher
生信软件10 - DNA/RNA/蛋白多序列比对图R包ggmsa
生信软件11 - 基于ACMG的CNV注释工具ClassifyCNV
生信软件12 - 基于Symbol和ENTREZID查询基因注释的R包(easyConvert )
生信软件13 - 基于sambamba 窗口reads计数和平均覆盖度统计
生信软件14 - bcftools提取和注释VCF文件关键信息
生信软件15 - 生信NGS数据分析强大的工具集ngs-bits



















 493
493

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











