






文章信息

发表期刊:Nature Communications
影响因子:14.7
研究背景
前列腺癌是男性常见的恶性肿瘤,尽管局部肿瘤预后较好,但转移性疾病的高死亡率及治疗抵抗仍是重大挑战。基因组与转录组研究已揭示了癌细胞的内在异常,但对肿瘤微环境(TME)的动态作用仍知之甚少。TME通过细胞间相互作用参与肿瘤进展、治疗响应和转移,但传统单细胞RNA测序(scRNA-seq)缺乏空间信息,难以解析TME的复杂空间组织。
本文聚焦于前列腺癌中一类特殊上皮细胞——club-like细胞。这类细胞最初在肺部被发现,近期研究表明其在前列腺外周区及炎症区域富集,且转录特征与小鼠去势抵抗性前列腺癌的管腔祖细胞相似。研究团队整合了120例患者的单细胞和空间转录组数据,覆盖良性增生、初治肿瘤、新辅助治疗后肿瘤及去势抵抗性前列腺癌(CRPC),系统解析了TME的时空动态变化。
技术路线
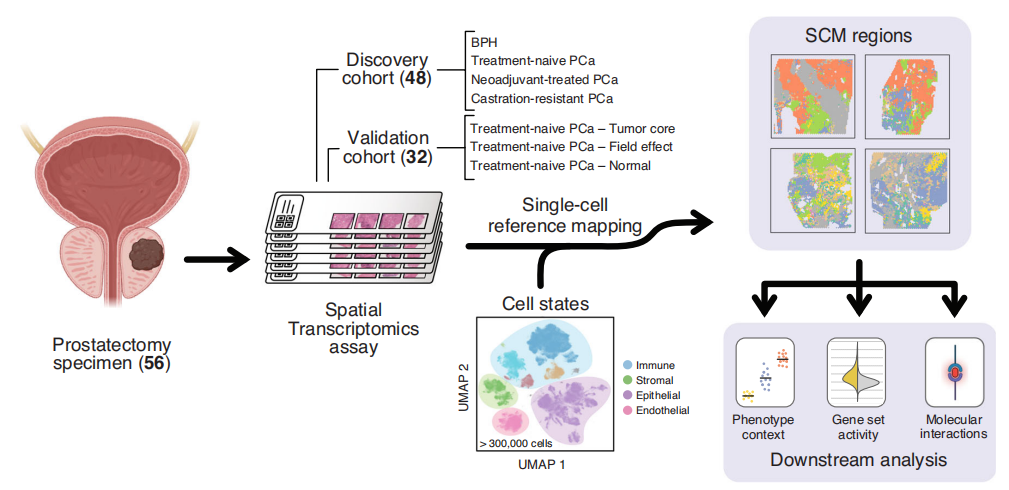
研究结论
本研究通过对单细胞和空间转录组学整合分析,揭示Club-like细胞作为前列腺癌肿瘤微环境(TME)的关键上皮亚型,其富集区域表现出雄激素信号耗竭、管腔祖细胞标记上调及衰老相关分泌表型(SASP),并通过高表达中性粒细胞趋化因子(如 CXCL1/2/8)显著促进多形核髓源抑制细胞(PMN-MDSC)浸润,形成免疫抑制微环境。该细胞亚群对雄激素剥夺治疗(ADT)具有耐药性,其介导的 PMN-MDSC 活性在原发及转移性肿瘤中均与免疫抑制密切相关,且通过配体 - 受体相互作用(如 CXCL2-ACKR1、CCL20-CCR6)与周围细胞通讯。该研究首次系统揭示了Club-like细胞通过协调免疫抑制性微环境驱动前列腺癌治疗抵抗的机制,为靶向TME的精准治疗(如联合免疫疗法与ADT)奠定了理论基础,同时提供了可转化的生物标志物和干预靶点。
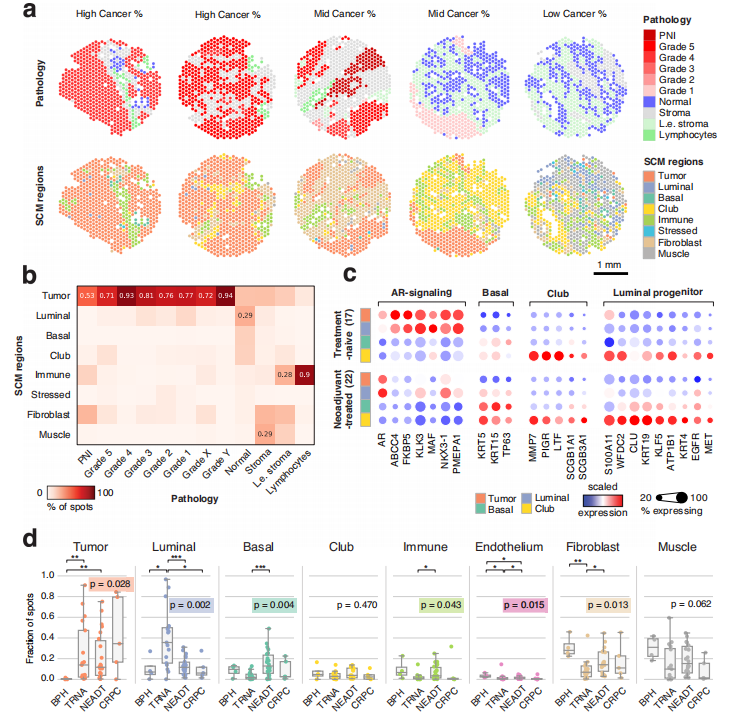
图1、单细胞映射衍生区域(SCM 区域)与组织病理学特征相符,并且反映了基因表达随治疗变化的情况。
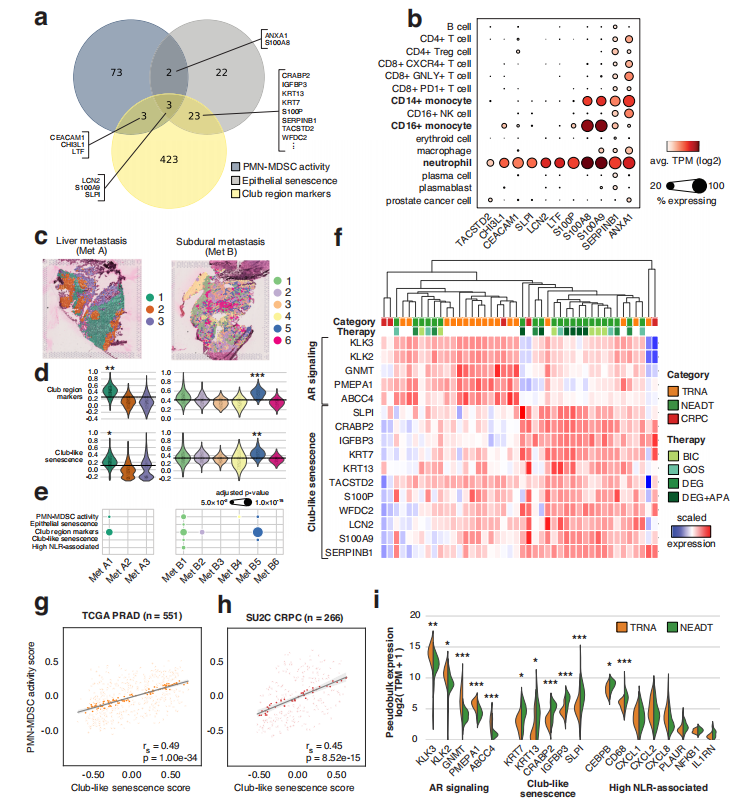
图2、在原发性和转移性肿瘤中,club-like细胞衰老与免疫抑制性多形核髓源抑制细胞(PMN-MDSC)的活性相关

















 960
960

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









