







 文章介绍了单细胞转录组分析中的细胞亚群细化过程,包括如何使用R包Seurat进行数据处理和聚类,以及如何根据细胞比例变化进行个性化分析。通过实例展示了如何识别并进一步划分细胞亚群,如巨噬细胞的M1和M2表型,并利用特征基因进行细胞功能注释和可视化。
文章介绍了单细胞转录组分析中的细胞亚群细化过程,包括如何使用R包Seurat进行数据处理和聚类,以及如何根据细胞比例变化进行个性化分析。通过实例展示了如何识别并进一步划分细胞亚群,如巨噬细胞的M1和M2表型,并利用特征基因进行细胞功能注释和可视化。
通常情况下,单细胞转录组拿到亚群后会进行更细致的分群,或者看不同样本不同组别的内部的细胞亚群的比例变化。
这就是个性化分析阶段,这个阶段取决于自己的单细胞转录组项目课题设计情况,我们了解到的各式各样的分析点,并不是通用的。比如如果要比较细胞亚群比例,就必须要有多个样本,如果是单个样本,其分析的内容及方法也会不尽相同,类似的问题以及涉及到的分析很多。
今天小编带大家来学习下“单细胞个性化分析之细胞亚群继续分群”,相信大多数单细胞转录组的小伙伴都能用到。
细胞亚群进一步划分原则
理论上细胞亚群是可以无限划分的,因为世界上没有两个一模一样的细胞,关键是要把握一个度,什么样的差异可以判定为不同细胞亚群,什么样的差异是可以容忍的细胞类群内部异质性。有一个策略就是找出主要因素和次要因素。主要因素划分为主要亚群,比如外周血里面的T,B细胞当然是不同亚群,但是T细胞里面还可以继续划分:CD4或者CD8的T细胞,甚至继续划分, 如下图所示:
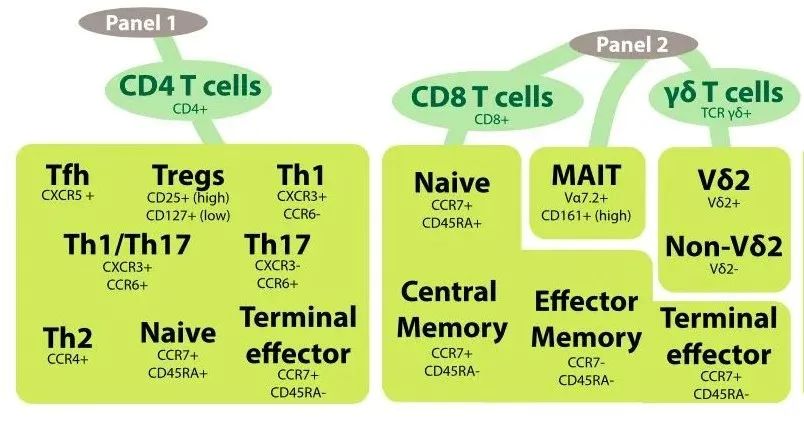
亚群细化分析实操
加载R包
{
library(Seurat)
library(dplyr)
library(reticulate)
library(sctransform)
library(cowplot)
library(ggplot2)
library(viridis)
library(tidyr)
library(magrittr)
library(reshape2)
library(readxl)
library(stringr)
library(cowplot)
library(scales)
library(readr)
library(progeny)
library(gplots)
library(tibble)
library(grid)
library(rlang)
theme_set(theme_cowplot())
use_colors <- c(
Alveolar_Macrophages = "#6bAEd6",
`Monocyte-derived macrophages`= "#fff500",
Monocytes= "#FA9FB5",
`Myeloid dendritic cells`= "#DD3497",
Plasmacytoid_dendritic_c 最低0.47元/天 解锁文章
最低0.47元/天 解锁文章


















 797
797

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









