






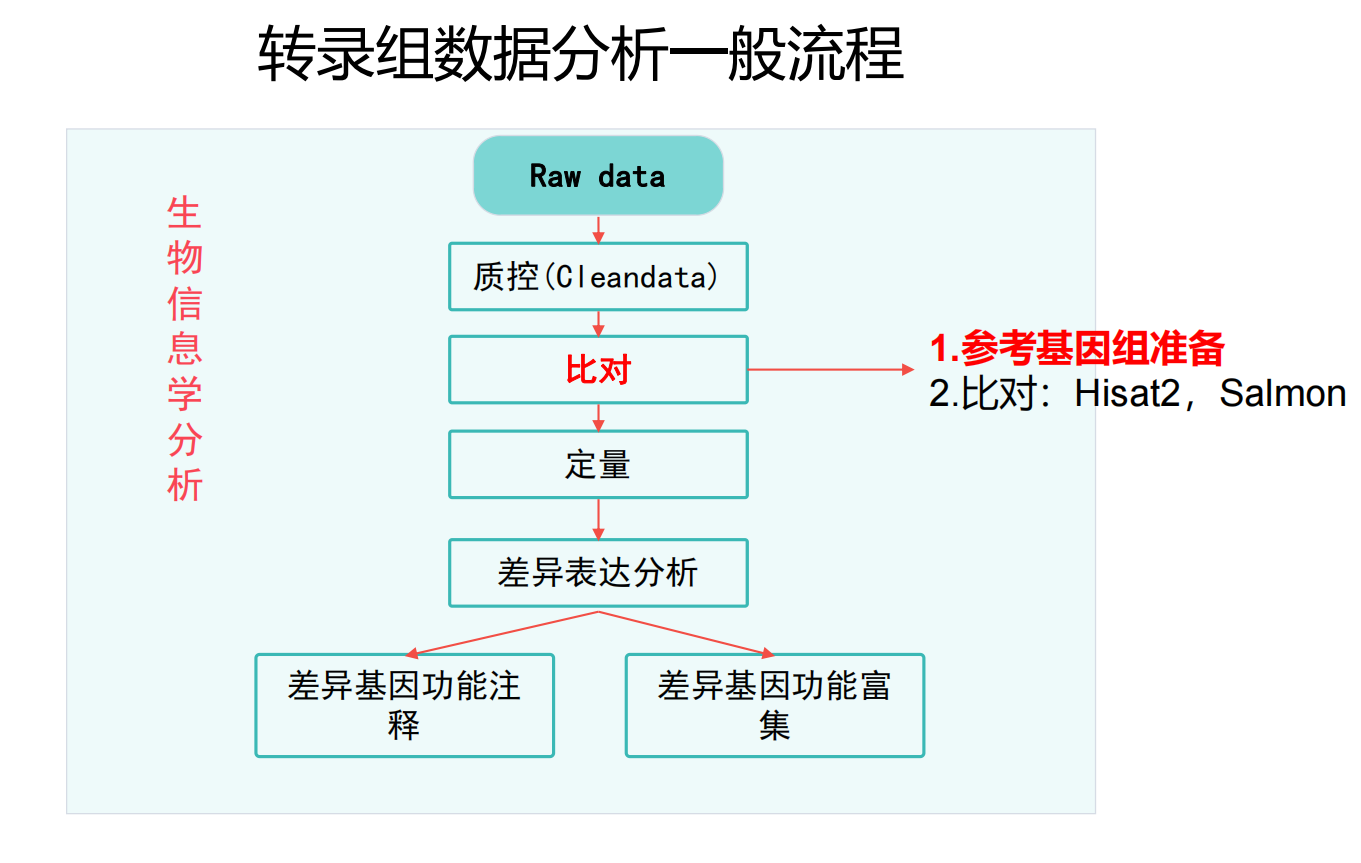
比对的流程:建立索引→比对到参考基因组→SAM转BAM文件→BAM建立索引
1.准备参考基因组、建立索引
## 参考基因组准备:注意参考基因组版本信息
# 下载,Ensembl:http://asia.ensembl.org/index.html
# http://ftp.ensembl.org/pub/release-104/fasta/homo_sapiens/dna/
# 进入到参考基因组目录
mkdir -p $HOME/database/GRCh38.105
cd $HOME/database/GRCh38.105
# 下载基因组序列axel curl
nohup wget -c http://ftp.ensembl.org/pub/release-105/fasta/homo_sapiens/dna/Homo_sapiens.GRCh38.dna.primary_assembly.fa.gz >dna.log &
# 下载转录组序列
nohup wget -c http://ftp.ensembl.org/pub/release-105/fasta/homo_sapiens/cdna/Homo_sapiens.GRCh38.cdna.all.fa.gz >rna.log &
# 下载基因组注释文件
nohup wget -c http://ftp.ensembl.org/pub/release-105/gtf/homo_sapiens/Homo_sapiens.GRCh38.105.chr.gtf.gz >gtf.log &
nohup wget -c http://ftp.ensembl.org/pub/release-105/gff3/homo_sapiens/Homo_sapiens.GRCh38.105.chr.gff3.gz >gff.log&
# 上述文件下载完整后,再解压;否则文件不完整就解压会报错
# 再次强调,一定要在文件下载完后再进行解压!!!
nohup gunzip Homo_sapiens.GRCh38.dna.primary_assembly.fa.gz Homo_sapiens.GRCh38.cdna.all.fa.gz >unzip.log &2.使用Hisat2比对到参考基因组上

输入:过滤之后的fastq.gz文件以及参考基因组目录
输出:sam文件
3.samtools
实现将sam文件转换为bam文件;对bam文件进行排序;为了能够快速访问bam文件,可以为已经基于坐标排序后bam或者cram的文件创建索引,生成以.bai或者.crai为后缀的索引文件。必须使用排序后的文件,否则可能会报错。另外,不能对sam文件使用此命令。
输入:sam文件
输出:sort.bam文件 bai文件
## ----构建索引
# 进入参考基因组目录
cd $HOME/database/GRCh38.105
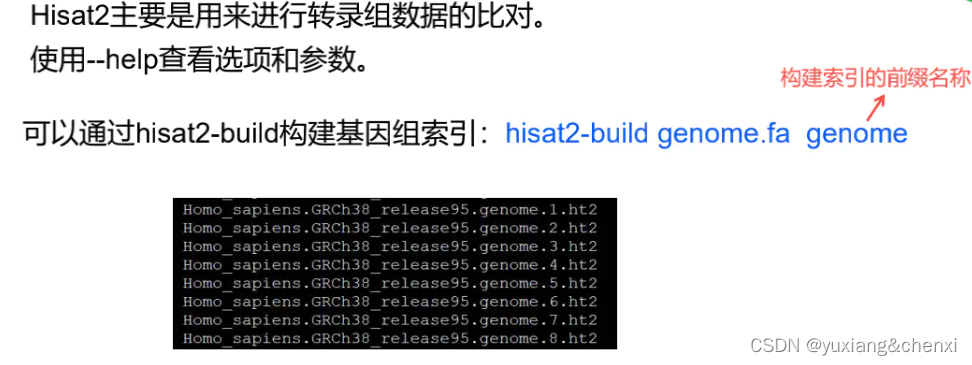
# Hisat2构建索引,构建索引时间比较长,建议提交后台运行,一般会运行20多分钟左右
## 后续索引可直接使用服务器上已经构建好的进行练习
vim Hisat2Index.sh
mkdir Hisat2Index
fa=Homo_sapiens.GRCh38.dna.primary_assembly.fa
fa_baseName=GRCh38.dna
hisat2-build -p 12 -f ${fa} Hisat2Index/${fa_baseName}
# 运行
nohup sh Hisat2Index.sh >Hisat2Index.sh.log &
## ----比对
# 进入比对文件夹
cd $HOME.../Mapping/Hisat2
## 单个样本比对,步骤分解
index=/home/t_rna/database/GRCh38.104/Hisat2Index/GRCh38.dna
inputdir=$.../data/cleandata/trim_galore/
outdir=$.../Mapping/Hisat2
hisat2 -p 10 -x ${index} \
-1 ${inputdir}/SRR1039510_1_val_1.fq.gz \
-2 ${inputdir}/SRR1039510_2_val_2.fq.gz \
-S ${outdir}/SRR1039510.Hisat_aln.sam
# sam转bam
samtools sort -@ 15 -o SRR1039510.Hisat_aln.sorted.bam SRR1039510.Hisat_aln.sam
# 对bam建索引
samtools index SRR1039510.Hisat_aln.sorted.bam SRR1039510.Hisat_aln.sorted.bam.bai
# 多个样本批量进行比对,排序,建索引
# Hisat.sh内容: 注意命令中的-,表示占位符,表示|管道符前面的输出。
## 此处索引直接使用服务器上已经构建好的进行练习
# vim Hisat.sh
index=/home/t_rna/database/GRCh38.104/Hisat2Index/GRCh38.dna
inputdir=$.../cleandata/trim_galore/
outdir=$.../Mapping/Hisat2
cat ../../data/cleandata/trim_galore/ID | while read id
do
hisat2 -p 5 -x ${index} -1 ${inputdir}/${id}_1_val_1.fq.gz -2 ${inputdir}/${id}_2_val_2.fq.gz 2>${id}.log | samtools sort -@ 3 -o ${outdir}/${id}.Hisat_aln.sorted.bam - && samtools index ${outdir}/${id}.Hisat_aln.sorted.bam ${outdir}/${id}.Hisat_aln.sorted.bam.bai
done
# 统计比对情况
multiqc -o ./ SRR*log
# 提交后台运行
nohup sh Hisat.sh >Hisat.log &b统计比对结果
# 进入比对文件夹
cd .../Mapping/Hisat
# 单个样本
samtools flagstat -@ 3 SRR1039510.Hisat_aln.sorted.bam
# 多个样本
ls *.sorted.bam | while read id
do
echo "samtools flagstat -@ 10 ${id} > ${id/bam/flagstat} "
done >flagstat.sh
# 运行
nohup sh flagstat.sh >flagstat.log &
# 质控
multiqc -o ./ *.flagstat

















 4万+
4万+

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









