







samtools depth命令简介
depth命令计算每一个位点或者区域的测序深度并在标准显示设备中显示。使用此命令之前必须先index。
命令格式:
samtools depth [options] [in1.bam|in1.sam|in1.cram[in2.bam|in2.sam|in2.cram]…]
参数:
-a 输出所有位点,包括零深度的位点;
-a –a,--aa 完全输出所有位点,包括未使用到的参考序列;
-b FILE 计算BED文件中指定位置或区域的深度;
-f FiLE 使用在FILE中的指定bam文件;
-I INT 忽略掉小于此INT值的reads;
-q INT 只计算碱基质量值大于此值的reads;
-Q INT 只计算比对质量值大于此值的reads;
-r CHR:FROM –TO 只计算指定区域的reads;
depth命令运行如下图所示:
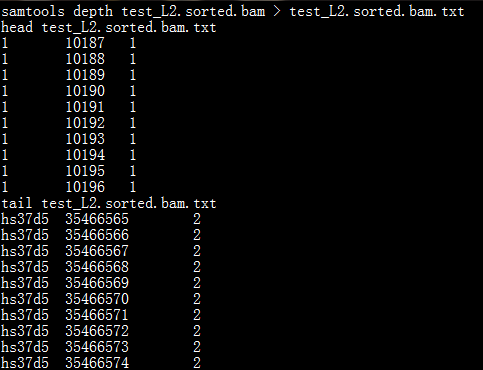
一共得到3列以指标分隔符分隔的数据,第一列为染色体名称,第二列为位点,第三列为覆盖深度。















 7997
7997

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









