






欢迎关注”生信修炼手册”!
基于全基因组数据来检测CNV是非常有效的一个手段,然而全基因组的成本还是挺高的。全外显子组在检测SNP方面已经比较成熟,考虑到外显子上的变异可能更具有致病性,科研人员也希望通过检测外显子上的CNV来实现一个高效,经济的CNV检测,很多的软件被开发用于WES的CNV分析。
CNV区域的长度可能横跨了多个外显子或者基因,断裂点位于外显子以外的位置,所以全基因组分析中Read-pair, split-read的策略无法应用到WES的CNV分析中,只能通过read-depth的策略来进行分析。
然而和全基因组不同,全外显子靶向捕获了基因组的外显子区域,考虑到GC含量,序列捕获等系统误差,其测序深度的分布和CNV之间的相关性更加复杂,建模衡量的难度更大,所以之前适用于WGS分析的CNV检测软件很多都不可以用于WES的分析。
为了有效减少系统误差的影响,提高CNV检测的准确率,很多WES的分析软件都会需要一个对照样本,将对照样本和测试样本进行比较来识别二者间差异的地方,从而回避系统误差带来的影响。同样的protocol意味着同样的系统误差,而二者直接还存在的差异就是由于样本本身的差异引起的了,这就是对照样本的作用。所以WES的CNV检测经典的用处就是检测体细胞CNV,即SCNA变异,提供配对的癌和癌旁样本来进行分析。
在以下文献中,详细列举了几种外显子CNV检测的软件
https://academic.oup.com/bib/article/16/3/380/245577
根据是否需要对照样本分成以下3大类
paired data, 需要配对的对照样本
pooled data, 不需要对照样本
paired and pooled data, 两种策略都可以
1. paired data
软件列表如下
ExomeCNV
Varscan2
Control-Freec
exome2cnv
PropSeg
2. pooled data
软件列表如下
condex
exomeCOPY
cn.mops
conifer
ExomeDepth
XHMM
ExoCNVTest
Excavator
3. paired and pooled data
软件列表如下
contar
ADTEx
FishingCNV
该文章发表于2014年,在之后又陆续发表了很多新工具,比如excavator, 2016年发表在Nucleic Acids Research上的文章介绍了excavator2进行CNV分析的强大之处,链接如下
https://academic.oup.com/nar/article/44/20/e154/2607979
不同工具算法模型都各不相同,各有优劣,在2014年发表的一篇文章对多个软件进行了评估,标题如下

在文章中,列举了很多CNV分析的软件,示意如下
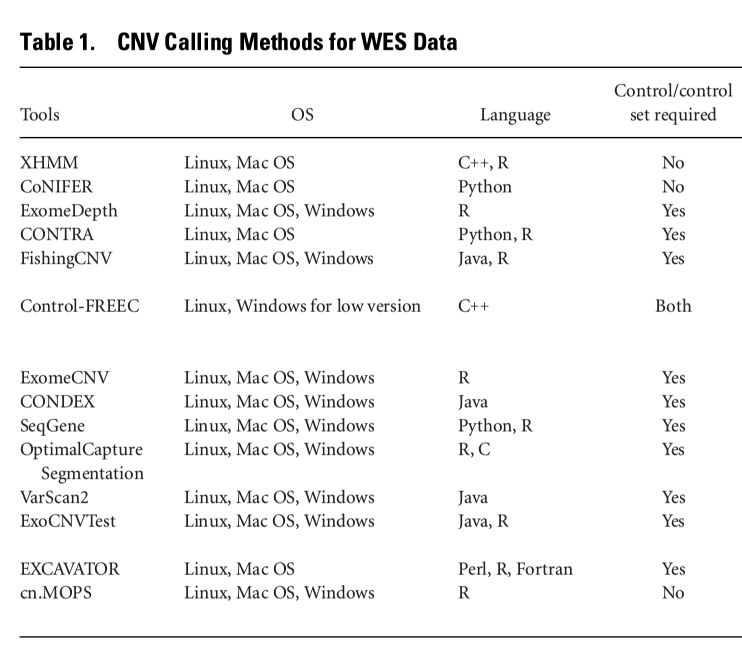
最终选取了以下4款软件来进行评估
XHMM
CoNIFER
ExomeDepth
CONTRA
从以下多个方面进行了评估
1. CNV长度和分布
不同软件检测到的CNV长度分布不同,结果统计如下
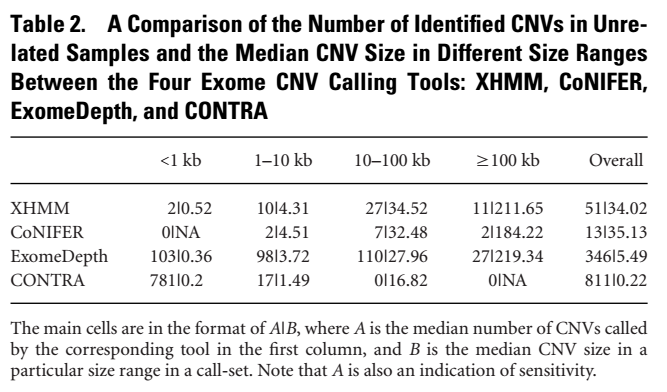
CNV的长度可以从几十bp跨越到几Mb的范围,通常认为小于300bp和长度在6kb左右的CNV应该是数量最多的。WES的CNV检测工具都是基于read-depth算法,采用滑动窗口的方法,窗口越大,最终鉴定出来的CNV可信度越高,所以在检测小片段的CNV方面,能力较差。
从统计结果可以看出,Conifer没有鉴定出1kb以下的CNV, 因为这款软件要求CNV至少需要覆盖3个exon区域,XHMM和ExomeDepth则可以同时检测小片段和大片段的CNV, CONTRA检测出来的数量过多,是由于其校正read-depthh的算法过于敏感,所以鉴定出来的CNV过多,在检测小于1kb的小片段CNV时,比较适合。
不同软件鉴定到的CNV的数量和类型展示如下
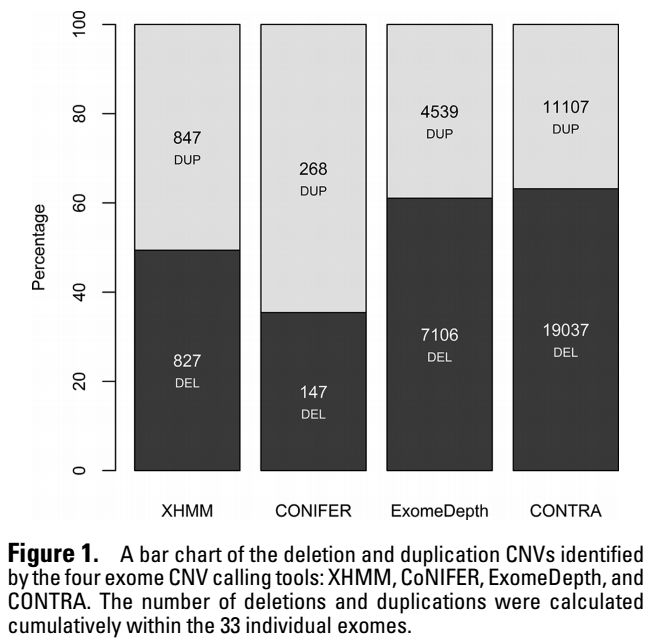
2. 和WGS的一致性
采用了cnvnator和ERDS两款软件对WGS数据进行CNV检测,然后和WES的结果进行一致性分析,以exon为单位进行评估,当一个exon 50%以上的区域落在CNV区域时进行计算,比较不同软件检测到的exon和WGS数据exon的overlap情况,结果如下

尽管都很低,但是很明显ExomeDepth overlap率最高,接下来是XHMM。
3. 和Common CNV的一致性
利用1000G项目中在人群中频率大于5%的cnvs作为common cnv, 采用上述的方法评估不同软件和common cnv的一致性,结果和WGS一致,也是ExomeDepth最高,XHMM次之。
4. Mendelian Error Rate评估
通常情况下denovo CNV的概率是非常低的,将denovo CNV作为Mendelian Error Rate的指标,对个体及其双亲同时进行CNV分析,评估denovo cnv的频率,结果如下
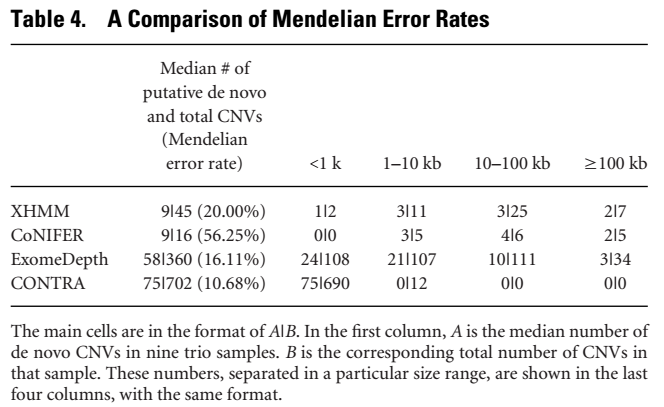
每个软件不符合孟德尔遗传的CNV比例都很高,conifer最高,而CONTRA最低。
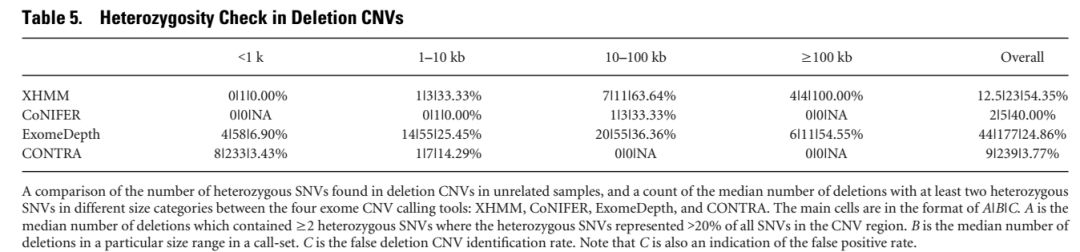
5. deletion CNV的假阳性检测
对于deletion CNV而言,其染色体区域只剩下一份拷贝,在该区域内的SNV必然为纯合性的,所以将包含了杂合SNV的CNV区域作为假阳性的结果,考虑到SNP分型的准确率,将同时满足以下两个条件的缺失区域定义为假阳性的结果
包含了2个以上的杂合SNP
20%以上的SNP位点为杂合
拷贝数缺失的假阳性统计结果如下
6. 不同软件之间的一致性
基于exon水平来统计不同软件之间的一致性,结果如下所示
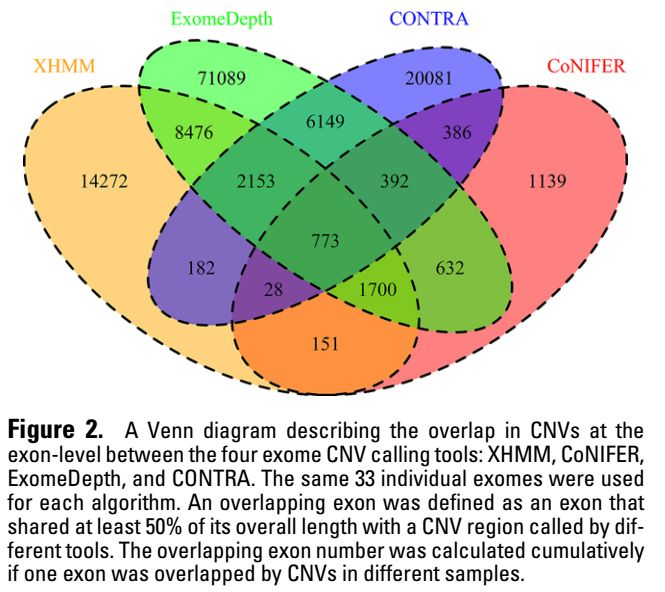
综合以上6个指标来看,没有哪个软件是全面优于其他软件的,在不同指标上,不同软件各有优劣。
在进行WES的CNV检测时,基于一款软件的结果很难兼顾灵敏度和特异性,最好的方法还是结合多款软件的结果进行判断。
·end·
—如果喜欢,快分享给你的朋友们吧—
扫描关注微信号,更多精彩内容等着你!


















 1145
1145

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









