







Genomics, Proteomics & Bioinformatics (GPB)发表了由深圳湾实验室系统与物理生物学研究所周耀旗课题组完成的题为“MARS and RNAcmap3: The Master Database of All Possible RNA Sequences Integrated with RNAcmap for RNA Homology Search”的论文。我们的“要文译荐”栏目很高兴邀请到作者团队为大家介绍MARS数据库的构建与RNAcmap3的应用。
研究问题
AlphaFold2 在蛋白质结构预测中的成功在相当程度上依赖于从蛋白质同源序列中获得的共进化信息,其同源序列提取自蛋白质序列的巨型整合数据库 Big Fantastic Database(BFD)。反观现有的核酸数据库群,尚缺乏为更广和更深的同源序列搜索整理的完备序列数据库。
研究方法
在NCBI nt及其分支数据库的基础上,通过整合来自 RNAcentral 的非编码RNA序列,来自 MG-RAST 的宏转录组和宏基因组序列,以及来自GWH和 MGnify 的基因组序列,构建完备的RNA序列集大成数据库 MARS,并针对巨型数据库改进RNAcmap同源序列搜索流程。
主要成果
本工作构建的 MARS 数据库序列规模约为 NCBI nt 数据库的20倍,或 RNAcentral 数据库的60倍。基于分卷搜索策略的新一代RNAcmap(RNAcmap3)在MARS上实现了超越当今最高水准技术的同源搜索表现。对于能够映射到 Rfam的有结构RNA,这一套新工具能为大多数目标序列获得比人工校对结果更精确和更敏感的多序列对齐结果。
MARS 简介
MARS 的序列数据整合自数个大型数据库的多种类型数据,包括NCBI 的核酸序列数据库 nt,env_nt,tsa_nt 和 patnt,非编码RNA序列数据库RNAcentral,宏基因组和宏转录组仓库MG-RAST,以及GWH和MGnify的基因组数据库。基因组中很大一部分都会转录为编码和非编码RNA,因此整合基因组序列可以最大限度地包含所有可能的RNA。NCBI 和 RNAcentral 提供的数据库发布前已经经过整理。MG-RAST,GWH,MGnify 积累的序列数据则由MARS团队按如下步骤整理(1)剔除长度超过1000Mb 的序列(通常是染色体序列),(2)所有序列以DNA字母表标准化,(3)序列中原有的空缺和非AT(U)GC 字母替换为“N”。经此处理后,以上序列数据连同NCBI 和 RNAcentral 数据以 fasta 格式粗合并的体积达到 1744 GB。粗合并的fasta 文件经 SeqKit 去除 100% 重复序列以后得到的 MARS 1.0 版本 fasta 文件体积达到 1571 GB,包含1,727,789,860 条核酸序列, 1,592,396,862,523 个碱基。MARS v1.0 可以从以下链接获取:https://ngdc.cncb.ac.cn/omix/release/OMIX003037

长按识别二维码获取链接
RNAcmap3 简介
RNAcmap3 是 RNAcmap 同源搜索流程的最新改进版本,其流程如图1所示。
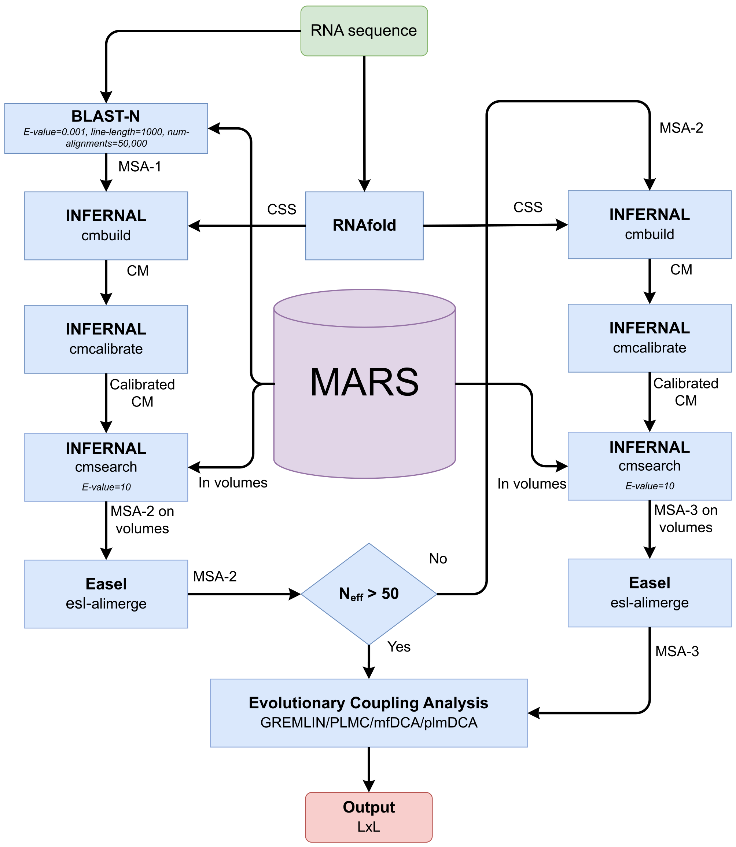
图1 RNAcmap3 流程示意图
RNAcmap的基本流程是:对于一个给定的RNA序列,首先调用 NCBI BLAST-N 从序列数据库(初版使用 NCBI nt)中生成一个初始MSA(MSA-1),并从一个单序列二级结构预测工具(如RNAfold)获得一致性二级结构(CSS)。利用 Infernal 从 MSA-1 和 CSS 构建共变量模型(CM)并根据该模型进行第二轮MSA搜索(MSA-2)以获得结果。RNAcmap的首个改进版本 RNAcmap2扩大了数据库规模到 nt 的分支序列库,并根据MSA-2中获得的有效同源序列个数,选择性地引入了在同源序列较少的MSA-2基础上重新构建CM并搜索MSA的第三轮循环(MSA-3)。在RNAcmap3中则进一步使用了巨型数据库 MARS,针对MARS数据量巨大的特点,在MSA-2和MSA-3轮搜索中采用了先分别搜索MARS分卷再合并结果的策略。RNAcmap2和RNAcmap3的改进均大幅提升了 RNAcmap 流程搜索RNA同源序列的能力。RNAcmap3 可以从以下链接获取:http://zhouyq-lab.szbl.ac.cn/download/

长按识别二维码获取链接
MARS & RNAcmap3 对 RNA 同源搜索的提升
同源搜索是展示MARS应用方向的好例子。在这个工作中,作者比较了使用MARS的RNAcmap3与两种当前最新的RNA 同源序列搜索工具:使用NCBI nt及其分支数据库的 RNAcmap2 和使用NCBI nt 和 RNAcentral 数据库的 rMSA, 对同一组基准RNA序列集进行同源搜索的表现。比较中各工具所使用的数据库的版本均与MARS 组成一致。作者对各工具搜索得到的MSA通过DCA共变量分析预测二级结构,并通过衡量预测二级结构的准确程度来评价各工具的同源搜索能力。用于测试的基准RNA序列集收集自RCSB PDB 并按80% 相似度去除序列重复性。得到的序列分别经 RNAcmap 对 NCBI nt 数据库搜索,并对获得的MSA进行评价,根据有效同源序列个数(number of effective homologous sequences, Neff)划分为四个同源丰富程度不同的集合:No-hit (Neff = 0), Low Neff (1 ≤ Neff < 10), Medium Neff (10 ≤ Neff < 50), 以及 High Neff (Neff ≥ 50),分别包含21,83,31和110条非冗余RNA序列。本工作的考察集中于No-hit, Low Neff 和 Medium Neff 集,因为对High Neff 序列,在使用 RNAcmap 时的DCA分析就已经达到了很高的准确程度。在衡量预测二级结构的准确程度时, 考察了对非局部碱基对(序号差大于3)预测的敏感性(sensitivity)SN = TP / (TP + FN),精确率(precision)PR = TP / (TP + FP), 和它们的调和平均数 F1 分数(F1-score)F1 = 2[PR × SN / (PR + SN)]。MSA 的共变量分析使用了四种DCA预测工具 GREMLIN, mfDCA, plmc 和 plmDCA。四种工具给出了相似的预测结果,其中以 mfDCA 的预测结果最佳,因此呈现的结果均使用mfDCA的预测。相关比较的结果如表1所示。
表1 RNAcmap2, RNAcmap3 以及 rMSA 的基准测试比较
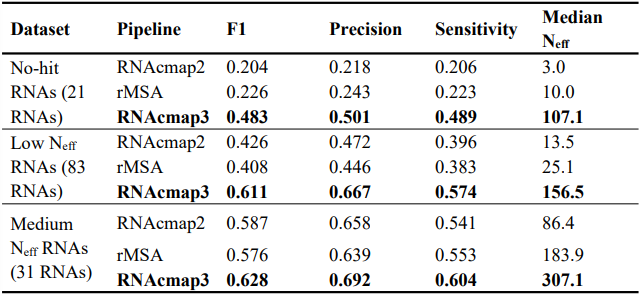
测试结果表明,RNAcmap2和rMSA在三个数据集上表现大致相当,RNAcmap3则在所有性能指标上超越了两者。平均来说 RNAcmap3 的 F1 分数比 RNAcmap2 在 No-hit 集上提升了136.8%, 在 Low Neff 集上提升了 43.4%, 在 Medium Neff 集上提升了 6.98%, 比 rMSA 则分别提升了 113.7%, 49.8% 和 9.0%。RNAcmap3 产生的 MSA 具有远高于 rMSA 和 RNAcmap2 的有效同源序列(Neff)数,这和从MSA获得的预测准确程度趋势是一致的。甚至,RNAcmap3在No-hit 集和 Low Neff 集上的表现分别优于 RNAcmap2在Low Neff 和 Medium Neff 集上的表现。RNAcmap3 在 Medium Neff 集上相对于 RNAcmap2或 rMSA的表现提升小于10%,因为在此序列集上后两者产生的MSA也具有了足够的Neff,这符合领域内的普遍认知:共变量分析的预测精度随MSA深度增加的提升存在上限。
RNAcmap3和人工注释的Rfam的比较
Rfam根据RNA的序列和二级结构同源性把RNA分类为家族。Rfam尽可能使用实验测定的二级结构进行同源搜索和对齐,而RNAcmap系列和rMSA 等方法使用 RNAfold 生成初始二级结构预测。因此,Rfam经常被认为是RNA MSA的金标准,即使并非Rfam中的所有RNA都使用了实验测定的二级结构。在本工作使用的RNA基准测试集中,有30条序列能够映射到30个Rfam 家族(1条No-hit,14条Low Neff,15条Medium Neff)。对这30个家族,用mfDCA分别利用来自RNAcmap3, RNAcmap2和Rfam的MSA 预测二级结构,结果列于表2。
表2 Rfam,RNAcmap2,RNAcmap3 和 RNAfold对Rfam和非Rfam映射 RNA的预测表现
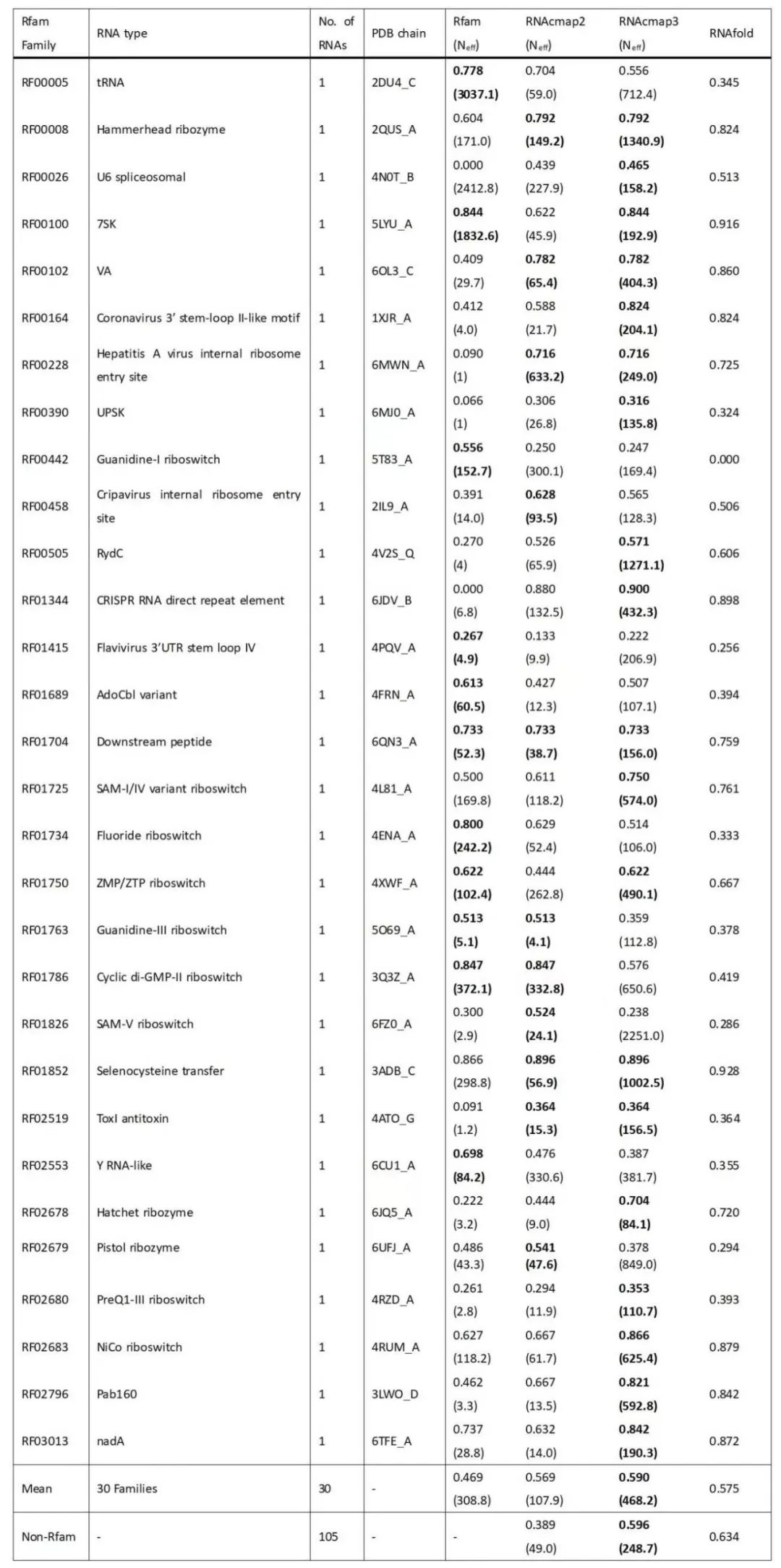
在17个家族中,RNAcmap3表现优于Rfam;在10个家族中 Rfam 表现优于 RNAcmap3;在3个家族中两者表现相当。RNAcmap 系列和Rfam的一个显著区别是,Rfam依赖已知的二级结构,而RNAcmap系列使用RNAfold预测的二级结构。RNAcmap系列 MSA的质量和RNAfold预测的种子二级结构的质量具有显著的正相关。对于RNAcmap3,其共变量分析得到的二级结构和RNAfold预测二级结构的全局Pearson相关系数达到了0.964,而RNAcmap2为0.470。这表明RNAcmap3找到的更多同源序列增强了MSA对RNAfold给出的种子二级结构的映射。另一方面,RNAfold 初始二级结构的预测质量也影响了RNAcmap3与Rfam比较时的表现,对于RNAfold 预测二级结构的F1分数大于0.51的家族,RNAcmap3的表现均不劣于 Rfam。
总结和展望
本工作建立了一个完备的核酸序列数据库MARS,拥有20倍于通常使用的nt数据库的核酸序列数。RNAcmap系列的最近改进版RNAcmap3通过在MARS上运用分卷搜索策略获得了更深的MSA 并获得了比RNAcmap2和rMSA 更好的共进化耦合。而且,尽管使用RNAfold预测的初始二级结构,RNAcmap3可以在很大程度上比Rfam MSA 导出更准确的二级结构。MARS和RNAcmap3将会为更深更广的RNA同源搜索做出贡献。
审校人:
GPB青年编委孙磊
文章编译来源:
Chen K, Litfin T, Singh J, Zhan J, Zhou Y. MARS and RNAcmap3: The Master Database of All Possible RNA Sequences Integrated with RNAcmap for RNA Homology Search. Genomics Proteomics Bioinformatics 2024. https://doi.org/10.1093/gpbjnl/qzae018.
英文全文详见:
https://academic.oup.com/gpb/advance-article/doi/10.1093/gpbjnl/qzae018/7617691?searchresult=1
作者信息:
周耀旗教授从2021年3月起全职加入了深圳湾实验室,他是1984年中国科技大学近代化学系的学士,1990年美国纽约州立石溪大学化学物理的博士,1994–2000年北卡州立大学、哈佛大学的博士后,2000年任纽约州立布法罗大学助理教授,2004年升为终身副教授,2006年成为印第安纳大学信息学院和医学院终身正教授,2013-2021年任澳大利亚格里菲斯大学糖组学研究所正教授。他长期在结构生物信息学方面工作,曾经多次在国际蛋白质/RNA结构预测和功能预测比赛中名列前茅。到目前为止共发表论文260余篇,引用1万7千多次,H因子72。目前,周耀旗课题组通过计算和实验的结合,从事蛋白质/RNA的序列、结构与功能关系方面的基础研究和生物分子检测、药物开发方面的应用研究。寻找在RNA/蛋白方面有AI计算、分子和细胞生物学实验相关经验的博士生和博士后。
GPB论文:
MARS and RNAcmap3: The Master Database of All Possible RNA Sequences Integrated with RNAcmap for RNA Homology Search
长按并识别二维码,阅读原文

相关推荐
GPB | RNA2Immune: 非编码RNA与免疫系统关联数据库
GPB | DeeReCT-TSS基于DNA序列和RNA-seq数据注释多种细胞类型中的转录起始位点
About GPB
Genomics, Proteomics & Bioinformatics(基因组蛋白质组与生物信息学报,简称GPB)于2003年创刊,是由中国科学院主管、中国科学院北京基因组研究所(国家生物信息中心)与中国遗传学会共同主办的英文学术期刊,由牛津大学出版社金色开放获取(Gold Open Access)出版。刊载来自世界范围内组学、生物信息学及相关领域的优质稿件。现为中国科学引文数据库(CSCD)和中国科技论文与引文数据库(CSTPCD)核心期刊,被SCIE、PubMed/MEDLINE、Scopus等数据库收录。2023年公布的官方数据显示,CiteScore为11.7;2年和5年Impact Factor分别为9.5和10.1,分别排名WoS遗传学领域12/171和13/171;2022 JCI为2.08,排名WoS遗传学领域10/189。期刊由科技部等七部门联合实施的“中国科技期刊卓越行动计划“资助(2019–2023)。

长按识别二维码
关注GPB

















 1035
1035

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









