






近年来,泛基因组学成为生物信息学领域中的一大热门研究方向。泛基因组指的是某一物种或种群的所有基因组序列,涵盖个体基因的存在和缺失情况。相比传统的基于单一参考基因组的方法,泛基因组学为我们提供了更全面的基因多样性视角,减少了参考基因组偏差,特别是在重测序项目中。美国加州大学Benedict Paten研究组在《Nature Biotechnology》上发表了一篇重要文章,介绍了一种名为Minigraph-Cactus的新工具。这一工具结合了minigraph的快速参考基因组到图的映射和Cactus的比对能力,为构建高质量的图形泛基因组提供了全新解决方案。本文将详细介绍Minigraph-Cactus的应用,特别是在构建和验证果蝇泛基因组中的表现。
泛基因组构建的背景与挑战
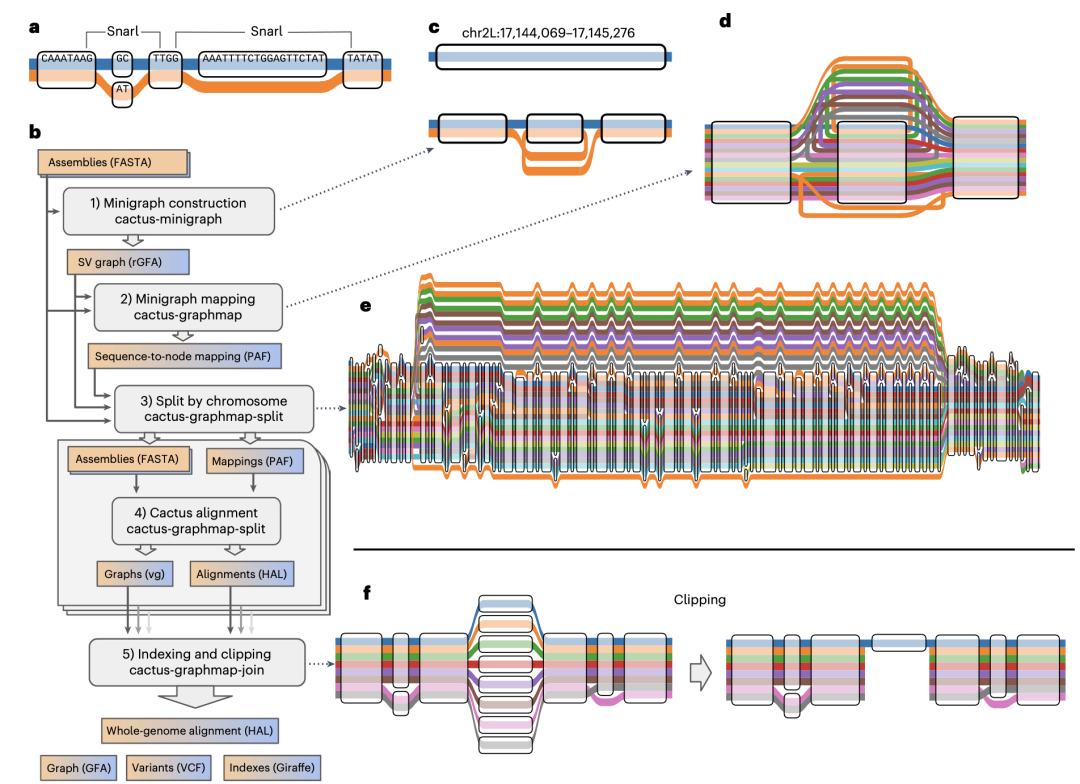
泛基因组的构建主要依赖于第三代测序技术的进步,使我们能够以高质量基因组为基础进行遗传变异研究。然而,基于单一参考基因组的方法存在显著缺陷。首先,依赖单一参考基因组可能导致偏差,尤其在重测序项目中;其次,展示结构变异的方式是一个重大挑战。为了应对这些问题,研究人员开发了图形化泛基因组及其对应的软件工具。
Minigraph-Cactus的创新点
Minigraph-Cactus是一种创新的泛基因组构建流程,结合了minigraph的快速参考基因组到图的映射和Cactus的比对能力,同时融入了vg中的几项关键改进。通过这一工具,研究人员成功构建了多个脊椎动物的图形泛基因组。这些图形泛基因组不仅能在各种分辨率下展示变异,还提升了短读和长读映射、变异调用及结构变异分型的研究水平。
Minigraph-Cactus泛基因组构建流程已经被集成到Cactus软件中,能够在本地和分布式计算集群上运行。该流程由五个步骤组成,可以单独运行或在单个工作流中一起运行。这些步骤生成的图形泛基因组以GFA格式和VCF格式输出,包括基因组比对结果和使用vg Giraffe进行映射所需的索引。
基于HPRC数据集的人类泛基因组构建
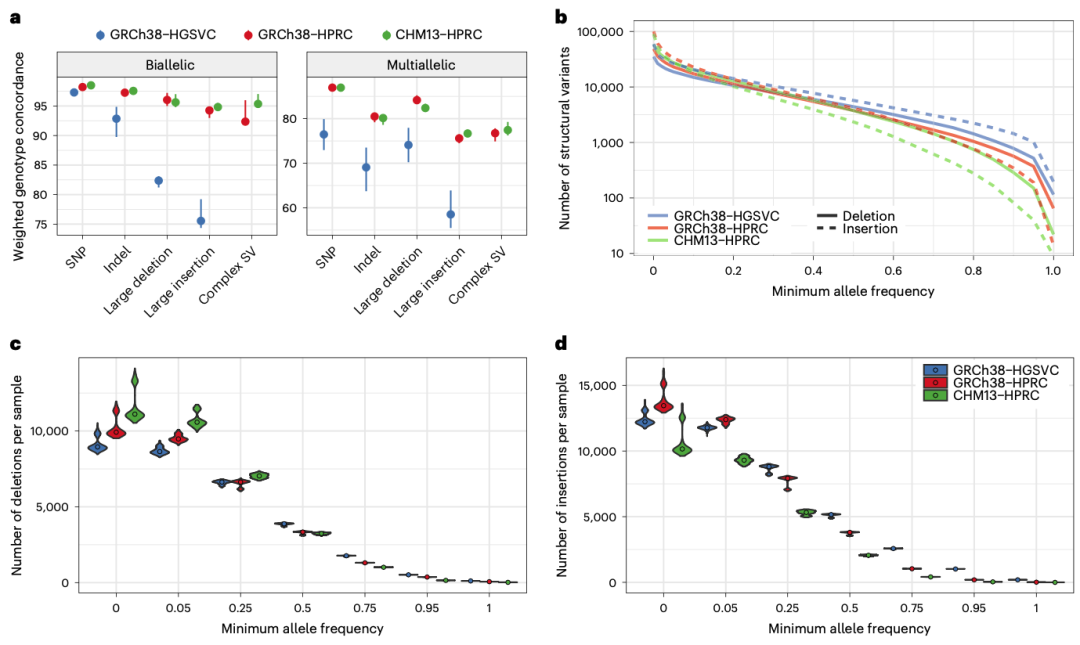
为了验证Minigraph-Cactus的效果,研究人员利用HPRC联盟发布的44个二倍体基因组数据,构建了一个包含90个单倍体基因组的图形泛基因组。在这一过程中,研究人员将GRCh38和CHM13(版本1.1)作为参考基因组进行比较。结果显示,基于CHM13的图形泛基因组包含更多高度变异区域,如chr21短臂上的复杂区域,使其比基于GRCh38的图形泛基因组略大。构建出的图形泛基因组比minigraph的结构变异多了约200倍的节点和边。图谱中非参考基因组中存在的序列数量随基因组数目的变化显著增加。
此外,研究人员还比较了VCF格式的图形泛基因组与Hi-Fi reads映射到GRCh38产生的变异,结果显示其精确度和重复性均很高。
PanGenie工具在结构变异基因分型中的应用
PanGenie是一种使用短读数据对人类结构变异进行基因分型的工具。它利用隐马尔可夫模型(HMM),结合来自泛基因组中已知单倍型的信息(VCF表示)与来自短读数据的k-mers进行基因型推断。研究人员通过对1000基因组计划中的368个样本进行基因分型来评估这一过程,包括随机选择的20个三联体及图谱中已有样本。研究结果显示,Minigraph-Cactus HPRC图形泛基因组在所有类型结构变异的准确性方面比人类基因组结构变异联盟(HGSVC)高得多。
果蝇泛基因组的构建与验证
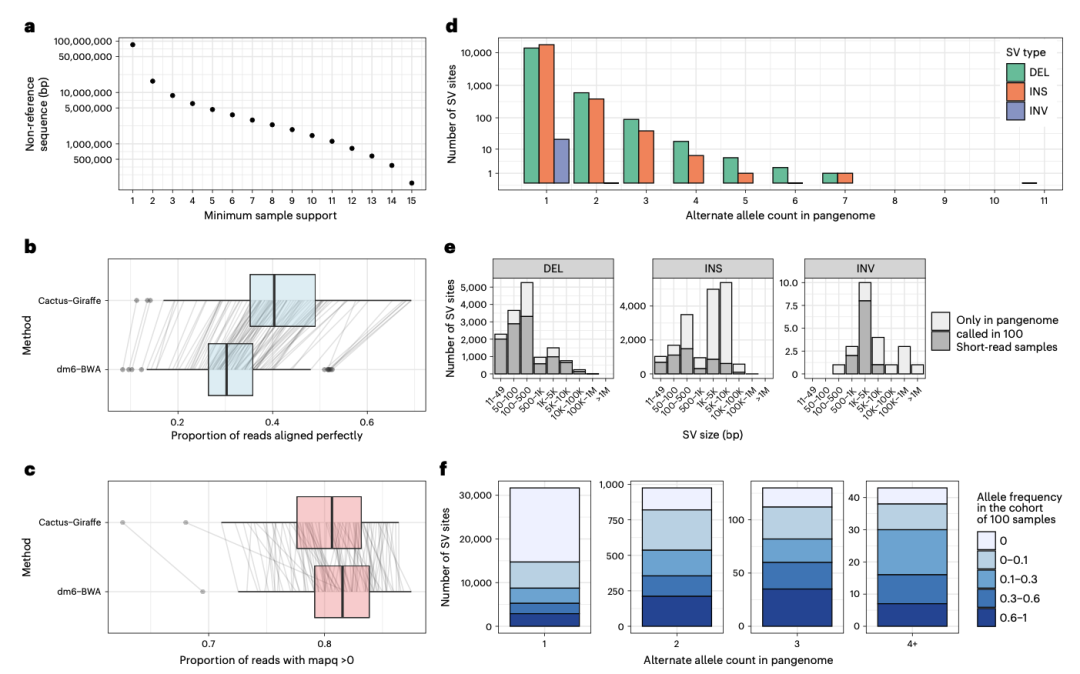
为了验证Minigraph-Cactus在非人类生物中的适用性,研究人员构建了果蝇泛基因组。使用了16个参考基因组,基因组大小范围从132Mb到144Mb,并构建了五个图形泛基因组。构建泛基因组大约需要5小时,而进行渐进式Cactus比对大约需要19小时。向结构变异图谱中添加base-level的变异会使节点和边的数量增加大约两个数量级。Minigraph-Cactus泛基因组的“核心”基因组大小为110Mb,大约占泛基因组总大小的一半。这表明样本之间具有较高的序列多样性,特别是转座子(TE)插入在果蝇中的丰富性。结果显示,非参考序列的数量随其存在的最小样本数的增加而增加。
结论与展望
总的来说,Minigraph-Cactus泛基因组构建流程为泛基因组研究提供了一种高效且准确的新方法。它能够直接从全基因组比对开始创建泛基因组,展示了其构建来自人类泛基因组参考联盟的90个人类单倍型泛基因组的能力。这一方法不仅能够构建包含所有形式遗传变异的图形化泛基因组,还可以使用基因分型工具进行基因分型。
研究人员还评估了参考基因组的质量和完整性对泛基因组构建的影响,表明使用T2T联盟的CHM13参考基因组提高了这一流程的准确性。最后,研究人员利用这一流程构建了果蝇的图形化泛基因组,验证了Minigraph-Cactus在非人类生物中的适用性。
Minigraph-Cactus的成功应用展示了图形化泛基因组在基因组学研究中的巨大潜力。未来,随着更多种类的基因组数据的积累和计算方法的进一步优化,这一工具有望在更加广泛的生物学研究中发挥重要作用。通过不断改进和推广,Minigraph-Cactus将推动泛基因组学的进一步发展,为揭示生物多样性和遗传变异提供更加全面和精准的视角。
详细数据与分析方法
在构建果蝇泛基因组时,研究人员使用了16个参考基因组,基因组大小范围从132Mb到144Mb。通过五个步骤,构建泛基因组大约需要5小时,而进行渐进式Cactus比对大约需要19小时。结构变异图谱中添加base-level的变异使节点和边的数量增加了两个数量级。Minigraph-Cactus泛基因组的“核心”基因组大小为110Mb,占泛基因组总大小的一半,表明样本之间具有较高的序列多样性。
使用的软件包括Minigraph-Cactus、Cactus、vg Giraffe等。数据分析通过集群上的分布式计算完成,生成的图形泛基因组以GFA和VCF格式输出。这些数据和方法的应用使得Minigraph-Cactus成为构建高质量泛基因组的理想工具。
总之,Minigraph-Cactus通过高效、准确的泛基因组构建流程,在揭示基因多样性和遗传变异方面展示了巨大的潜力。
# 快速开始脚本
# 创建模拟灵长类动物的泛基因组
cactus-pangenome ./js ./examples/evolverPrimates.txt --outDir primates-pg --outName primates-pg --reference simChimp --vcf --giraffe --gfa --gbz
# 检查输入序列是否作为contigs显示在GFA中
zcat primates-pg/primates-pg.gfa.gz | grep '^W' | awk '{print $1 "\t" $2 "\t" $3 "\t" $4 "\t" $5 "\t" $6 }' | more
# 检查参考路径,在header中识别出来
zcat primates-pg/primates-pg.gfa.gz | head -1
# 基于参考路径(simChimp)生成VCF,并为每个单倍型生成一个样本
gzip -dc primates-pg/primates-pg.vcf.gz | grep CHROM -A 1
# 生成giraffe所需的三个输入文件
ls -hs primates-pg/primates-pg.d2.*
# 构建泛基因组图
cactus-minigraph <jobStore> <seqFile> <outputGFA> --reference
# 将每个输入组装映射回图
cactus-graphmap <jobStore> <seqFile> <inputGFA> <outputPAF> --reference
# 可选步骤:拆分输入组装和PAF文件
cactus-graphmap-split <jobStore> <seqFile> <inputGFA> <inputPAF> --reference --outDir
# 计算多个基因组对齐
cactus-align <jobStore> <seqFile> <inputPAF> <outHal> --reference --pangenome --outVG --maxLen
# 生成最终图和索引
cactus-graphmap-join <jobStore> --vg --outDir --outName --reference
















 3423
3423

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









