






场景:有时处理芯片数据和TCGA数据会发现,不同数据的基因名不同。
原因:基因可能有多个名字
Entrez ID:为一串数字,是NCBI数据库给每一个基因指定的唯一ID,例如TP53的Entrez ID为7157
一个 Entrez ID 可能对应多个 Gene symbol,如TP53还有其他名字如P53、LFS1等
GPL的EntrezID与Symbol 提取
##GPL的探针处理
#EntrezID与Symbol 进行一一对应#
rm(list=ls())
GPL <- read.delim("GPL570.annot",stringsAsFactors=FALSE,
skip = 27 )
gpl <- GPL[,c(3,4)]
colnames(gpl) <- c("Gene_symbol","Gene_ID")
##进行去重
gpl1 <- gpl[!duplicated(gpl$Gene_ID),]##这个好像正确
dim(gpl1)
#[1] 22190 2
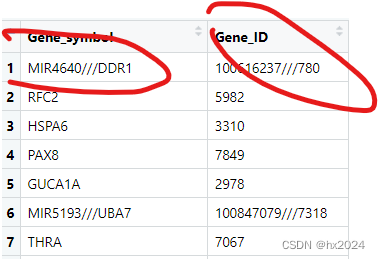
##对100616237///780中的///进行处理
##方法一直接写csv,然后进行Excel行列转换,然后进行分列
##方法二:使用代码#####################################
df1 <- as.data.frame(gpl1[,1])
new_df1 <- data.frame()
for (i in 1:nrow(df1)) { # 将每个元素按照"///"进行分割
elements <- unlist(strsplit(as.character(df1[i, 1]), "///")) # 将分割后的元素逐个添加到新的dataframe中
for (j in 1:length(elements)) { new_df1 <- rbind(new_df1, data.frame(V1 = elements[j])) } }
colnames(new_df1) <- "Gene_symbol"
df2 <- as.data.frame(gpl1[,2])
new_df2 <- data.frame()
for (i in 1:nrow(df2)) { # 将每个元素按照"///"进行分割
elements <- unlist(strsplit(as.character(df2[i, 1]), "///")) # 将分割后的元素逐个添加到新的dataframe中
for (j in 1:length(elements)) { new_df2 <- rbind(new_df2, data.frame(V1 = elements[j])) } }
colnames(new_df2) <- "Gene_ID"
data <- cbind(new_df1,new_df2)##去除"///"后的文件
dim(data)
##[1] 24563 2
#########################################
##进行数据检查,是否正确##
#gpl1 含有///
gpl2 <- data##单个数据
length(table(intersect(gpl1$Gene_symbol,gpl2$Gene_symbol)))
##[1] 20848(这些数据是否对应)
gene <- intersect(gpl1$Gene_symbol,gpl2$Gene_symbol)
gpl11 <- gpl1[gpl1$Gene_symbol%in%gene,]
dim(gpl11)
[1] 20848 2
gpl22 <- gpl2[gpl2$Gene_symbol%in%gene,]
colnames(gpl22)[2] <- "Gene_ID2"
gpltest <- merge(gpl11,gpl22,by="Gene_symbol")
identical(gpltest$Gene_ID,gpltest$Gene_ID2)
[1] TRUE##数据是一致的
##在检查///分隔的数据(肉眼。。。)
##没想到好的方法。。。
write.csv(gpl2,file = 'GPL570EntrezID_Symbol.csv')
结果:
ENSEMBL与Entrez ID对应
看R语言GeneID转换_ensg id转化 r语言_Joey_Liu666的博客-CSDN博客
参考:【精选】常见4种基因ID如何区别?如何转换?_gene symbol-CSDN博客
得到更多的人ENSEMBL与Entrez ID一文教你进行多种ID转换(上) - 知乎 (zhihu.com)
第一步从NCBI下载gene2ensembl文件:Index of /gene/DATA
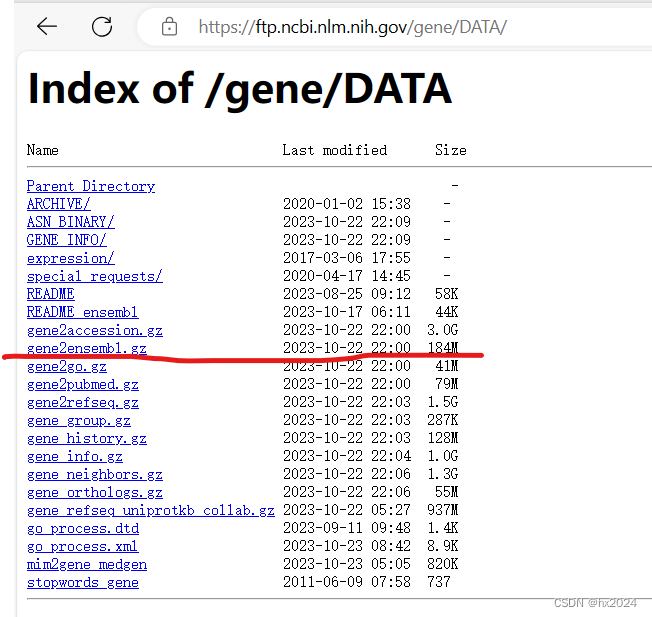
第二部R读取和简单的提取即可
#https://ftp.ncbi.nlm.nih.gov/gene/DATA/(下载数据)
rm(list=ls())
library(data.table)
gene2ensembl = fread("gene2ensembl.gz")##读取NCBI下载文件
gene2ensembl = data.frame(gene2ensembl,check.names = F)
colnames(gene2ensembl)[1] = "tax_id"
colnames(gene2ensembl)
#[1] "tax_id" "GeneID"
#[3] "Ensembl_gene_identifier"
#tax_id、GeneID和Ensembl_gene_identifier
##tax——id,为物种的ID,例如人类为9606
##"RNA_nucleotide_accession.version"NM编号(RefSeq 的转录本号
data <- gene2ensembl[,c(1:3)]
length(table(unique(data$tax_id)))
#[1] 503 个物种
datahs <- data[data$tax_id=="9606",]
write.csv(datahs,file = 'datahsEntrez_Ensembl.csv')##结果保存















 6678
6678











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









