









这个推文也在电脑里待了快一年了,拖延症患者,今天终于把它发出来了。NMDS分析过程已经R统计-PCA/PCoA/db-RDA/NMDS/CA/CCA/DCA等排序分析教程中写过了。最近又重新看了《Numerical Ecology with R》一书,巩固一下知识,正好重新整理了一下发出来。
目录:
一、数据准备
二、NMDS分析
2.1 vegan::metaMDS()
2.2 NMDS分析-labdsv::bestnmds()
三、绘制NMDS散点图
四、多元统计分析
五、拟合环境变量
5.1 线性拟合-envfit()
5.2 曲面拟合-ordisurf()或surf()
六、参考资料
一、数据准备
此数据为虚构,可用于练习,请不要作他用。
# 1.1 设置工作路径
#knitr::opts_knit$set(root.dir="D:\\EnvStat\\PCA")# 使用Rmarkdown进行程序运行
Sys.setlocale('LC_ALL','C') # Rmarkdown全局设置
#options(stringsAsFactors=F)# R中环境变量设置,防止字符型变量转换为因子
setwd("D:\\EnvStat\\PCA")
library(vegan)
library(ggplot2)
# 物种组成数据
## 原始计数
otu <- read.csv("otu.csv",header = TRUE ,row.names=1,sep = ",",stringsAsFactors = TRUE)
tail(otu)
dim(otu)
## 相对丰度
spe <- data.frame(otu[1:3],sweep(otu[-c(1:3)],1,rowSums(otu[-c(1:3)]),"/"))
tail(spe)
# 环境因子数据
env <- read.csv("env.csv",header = TRUE ,row.names=1,sep = ",",stringsAsFactors = TRUE)
tail(env)
dim(env)
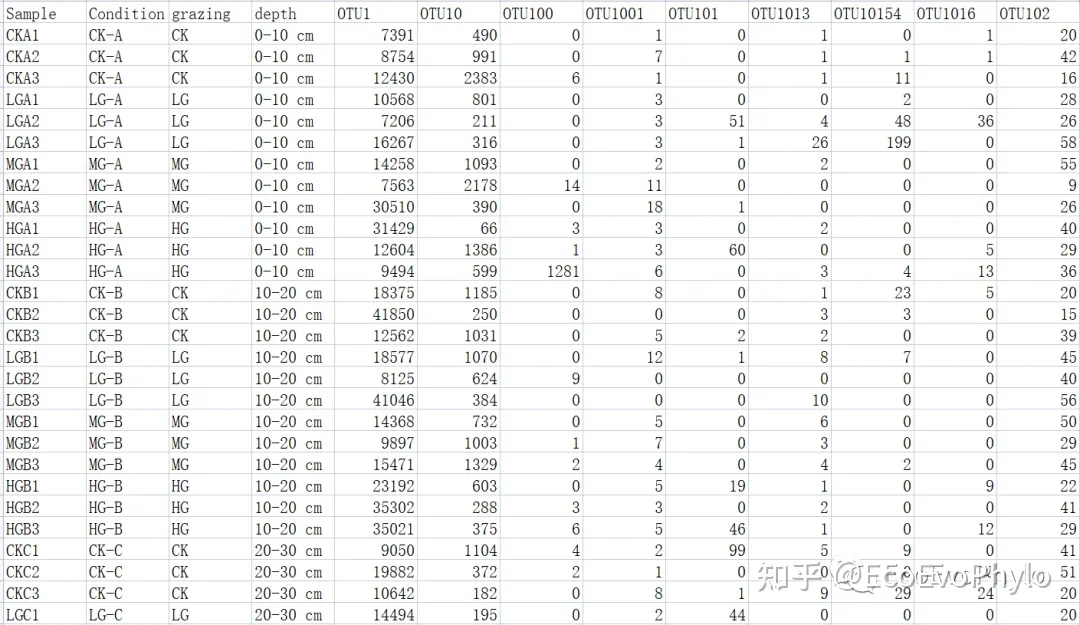
图1|otu及分组信息表,otu.csv。行为样品名称,列为otuID和分组信息。
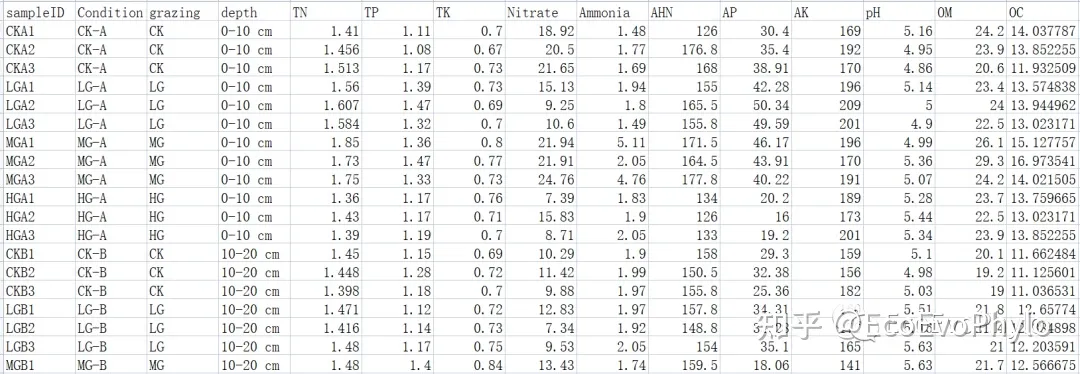
图2|环境因子及分组信息表,env.csv。行为样品名称,列为环境因子和分组信息。
二、 NMDS分析
在PCA、PCoA、CA和NMDS非约束排序方法中,只有NMDS不是基于特征向量的排序方法,不会最大化排序轴所代表的变量方差(可旋转轴,达到PCA的效果,PC1代表最大方差)。NMDS使用相异矩阵,沿预定数量的轴表示观测数据,同时保留观测间的排序关系。vegan::decostand()提供了一系列群落生态学的数据标准化方法。选择"standardize",可使变量的均值为0.方差为1。如果数据中存在较多的0,一般用hellinger。这里选择"total"计算otu相对丰度值,用于进行NMDS分析。有推文[NMDS分析](NMDS分析 - 知乎)提到“NMDS分析对0值不太敏感,即使有较多的0,也可以得到较为稳健的结果”。如果拿不准要选择那种转换方法和距离指数,就在满足分析假设的前提下,把所有分析都做一遍,选择分析效果更好的方法就行,不要让自己陷入纠结,浪费时间。
2.1 vegan::metaMDS()
# 2.1.1 NMDS分析
#??decostand # 查看数据标准化方法介绍。
#??vegdist # 查看可选的距离指数。
set.seed(12345)
pre1 <- cmdscale(vegdist(spe[-c(1:3)]), k = 2) # 也可使用metaMDS()的运行结果作为起始。
pre1 # 作为NMDS随机起始
set.seed(12345)
NMDS1 <- metaMDS(decostand(otu[-c(1:3)],method = "total"),# 等于spe[-c(1:3)]
# 调用vegdist()计算dissimilarity indices。designdist()还可以指定以相异指数。
distance = "bray",
engine = "monoMDS",# vegan还可以调用MASS的"isoMDS"进行NMDS分析。
trymax = 200,# 设置最大迭代次数,默认20。当出现2个收敛的解或达到最大迭代次数,NMDS分析停止。
wascores = TRUE, #wascores()计算变量得分,但数据中存在负值,则无法计算变量得分。
k=2, # 样本数需要大于2*k+1。
previous.best = pre1
)
NMDS1
# 2.1.2 输出结构介绍
#str(NMDS1)
NMDS1$iters # 迭代不到200,结果就收敛了,所以设置的trymax是足够的。
NMDS1$stress # 胁迫系数。
head(NMDS1$points)# 样本得分
head(NMDS1$species) # 若数据中存在负值,则无法计算出物种得分,所以最好选择数据转换后在[0,1]范围内的标准化方法。
## 若标准化后环境因子数据中存在负值,无法计算变量得分。
## 使用sppscores()可使用data原始数据计算变量得分,
## 也可使用不产生负值的标准化方法,如,Max等。
#sppscores(NMDS1) <- spe[-c(1:3)]
#NMDS1$species
# 2.1.3 Shepard diagram:goodness-of-fit,样本/观测点原始距离与NMDS排序结果比较。
pdf("stressplot.pdf",height = 6,width = 12)
gof <- goodness(NMDS1) # 值越大,观测在排序空间的位置与原始位置差距越大
par(mfrow=c(1,2))
stressplot(NMDS1,pch=16,p.col="black",l.col="red",main="Shepard plot") # R2 = 1-S*S
plot(NMDS1, display="site",type="t",main = "Goodness of fit")
points(NMDS1, display="site",col="red",cex=gof*100) # 气泡越大,观测在排序空间的位置与原始位置差距越大。
dev.off()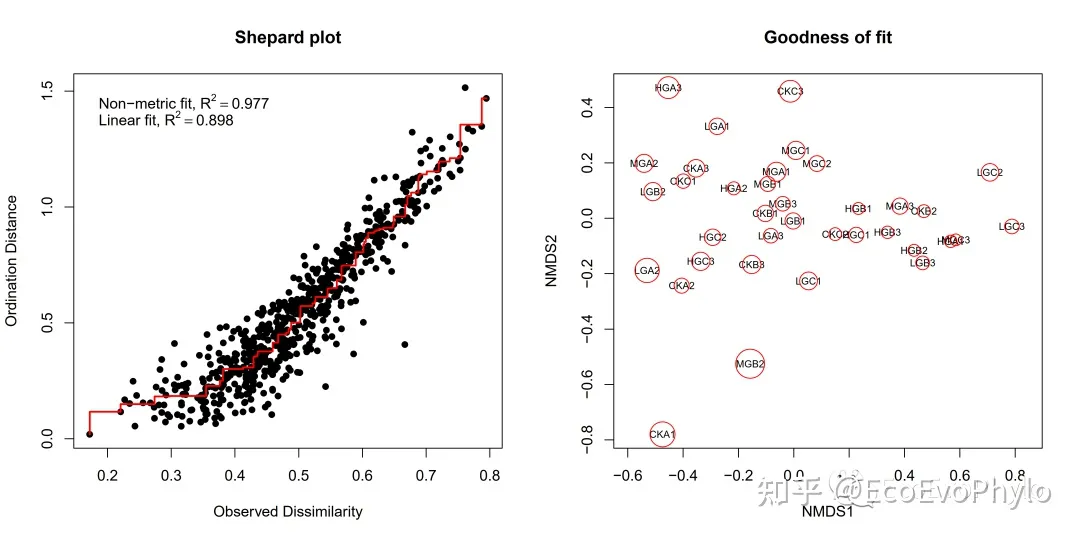
图3|Shepard diagram。样本/观测点原始距离与NMDS排序结果比较。胁迫系数表示原始差异的经NMDS排序后产生的变化,变化越小,表示NMDS排序效果越好,越能准确反应样本的原始空间位置或梯度。Non-metric fit R^2=1-stress^2。拟合优度图(右侧)中气泡越大,观测在排序空间的位置与原始位置差距越大。
# 2.1.4 数据提取
## 提取样本坐标数据
points1=NMDS1$points
head(points1)
## 提取物种坐标数据
Species1=NMDS1$species
head(Species1)
## 提取胁迫系数:表示原始差异的变异
stress1=round(NMDS1$stress,4) # 保留4位小数点
stress1 # 0.1517
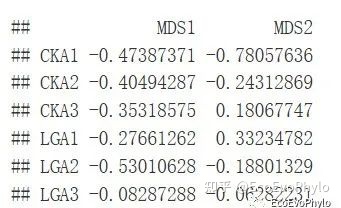
图4|样本得分,points1。
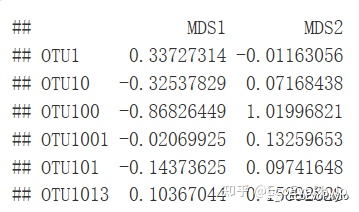
图5|物种得分,species1。
2.2 NMDS分析-labdsv::bestnmds()
bestnmds是使用isoMDS()进行NMDS分析,自动使用cmdscale()进行PCoA分析,作为初始随机起始值。
# 2.2.1 NMDS分析
library(labdsv)
dist <- vegdist(spe[-c(1:3)],method = "bray")
set.seed(12345)
NMDS2 <- labdsv::bestnmds(dist,
k=2,
itr=50,# 使用PCoA的结果作为初始随机起始值。
maxit=200,trace=FALSE)
NMDS2
isoMDS()的胁迫系数以百分比形式显示,monoMDS则以分数形式展示。
# 2.2.2 提取分析结果
## 样本坐标
points2 <- matrix(NMDS2$points, nrow = 36, ncol = 2,
dimnames = list(rownames(spe),c("MDS1","MDS2")))
head(points2)
## 物种得分果-不自动计算物种得分
vare.points <- postMDS(points2, dist) #看函数是对数据进行了PCA旋转,具体的可以自己看函数代码,postMDS.R。
head(vare.points)
species2 <- wascores(vare.points, spe[-c(1:3)], expand=FALSE)
head(species2)
### 可以直接使用NMDS2结果计算
species2.2 <- wascores(points2, spe[-c(1:3)], expand=FALSE)
head(species2.2)
#eigengrad(vare.points, spe[-c(1:3)])
## 绘图-两者数值范围不同,但相对位置差不多,上下,左右颠倒了位置。
pdf("bestnmds.species.pdf",height = 6,width = 12,family = "Times")
par(mfrow=c(1,2))
plot(species2)
text(species2,rownames(species2),cex=0.5)
plot(species2.2)
text(species2.2,rownames(species2.2),cex=0.5)
dev.off()
## 提取胁迫系数:表示原始差异的变异
stress2=round(NMDS2$stress,2) # 保留2位小数点
stress2 # 15.21%,即0.1521

图6|bestnmds样本得分,points2。
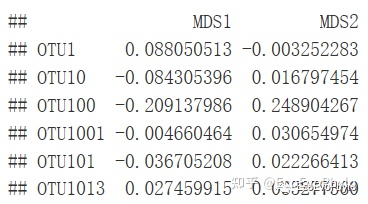
图7|bestnmds物种得分,species2。
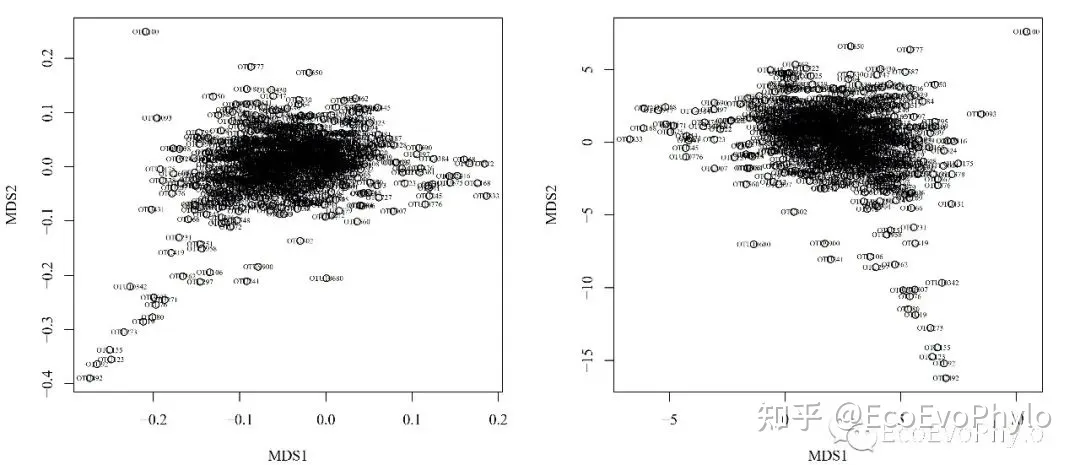
图8|不同方法计算物种得分的差异。两者数值范围不同,但相对位置差不多,上下或左右颠倒了位置。
三、 绘制NMDS散点图
图就是简单的散点图,会着重介绍一下,如何自定义图例。将两个分类因子的图例合并。
# 3.1 样本分组信息与样本坐标信息合并
mydata1=cbind(points1,spe[match(rownames(points1),rownames(spe)),1:3])
mydata1
mydata2=cbind(points2,spe[match(rownames(points2),rownames(spe)),1:3])
mydata2# 3.2 自定义图例-用不同颜色展示grazing,depth用颜色深浅(alpha)表示。
library(ggsci)
library("scales")
## 3.2.1 颜色选择
mypal = c(pal_d3(palette="category10",alpha = 0.2)(4),
pal_d3(palette="category10",alpha = 0.6)(4),
pal_d3(palette="category10",alpha = 1)(4)
)
show_col(mypal) # 查看颜色
mypal
## 3.2.2 绘图
p1 <- ggplot(data=mydata1,aes(MDS1,MDS2))+
geom_point(data=mydata1,
aes(color=Condition),
size=4.5,shape=16)+ # size可以修改点大小。
scale_x_continuous(limits = c(-0.8,0.8),breaks = seq(-0.8,0.8,0.2))+
scale_y_continuous(limits = c(-0.8,0.8),breaks = seq(-0.8,0.8,0.2))+ # scale*设置坐标区间。
annotate("text",x=0.7,y=0.7,size=5.75,colour = "black",# x,y设置stress值的放置位置。
label="paste('Stress=',0.15)",parse = TRUE)+
scale_color_manual(values = mypal,
breaks = c( # 自定义分类变量顺序,与颜色顺序对应。
"CK-A","LG-A","MG-A","HG-A",
"CK-B","LG-B","MG-B","HG-B",
"CK-C","LG-C","MG-C","HG-C"
),
labels = c(
"CK","LG","MG","HG",
rep("",8)
),
guide = guide_legend(
title=paste("0-10 cm","10-20 cm","20-30 cm",sep = "\n\n\n"),
direction = "horizontal",
title.position = "left",
title.hjust = 0, # 标题水平偏移
title.vjust = 0, # 标题垂直偏移
label.position = "top", # 设置标签放置位置
#label.hjust =0, # 设置图例标签左对齐
keyheight = 1, # 图例高度
keywidth = 2, # 图例宽度
ncol=4,
byrow = TRUE)
)+
theme_bw()+
labs(x="NMDS1",y="NMDS2")+
theme(# 调整图细节:轴标题,刻度字体大小等。
panel.background = element_blank(),
panel.grid = element_blank(),
axis.title = element_text(size=18,color = "black"),
axis.text = element_text(size=16,color = "black"),
legend.text = element_text(size=16,color="black"),
legend.title = element_text(size=16,colour = "black"),
legend.position= "right"
)
p1
## 3.2.3 输出pdf格式图片到本地
pdf("p1.pdf",height = 6,width = 10,family = "Times")
print(p1)#输出图的depth需自己对齐一下。
dev.off()
图9|设置的渐变颜色,mypal。
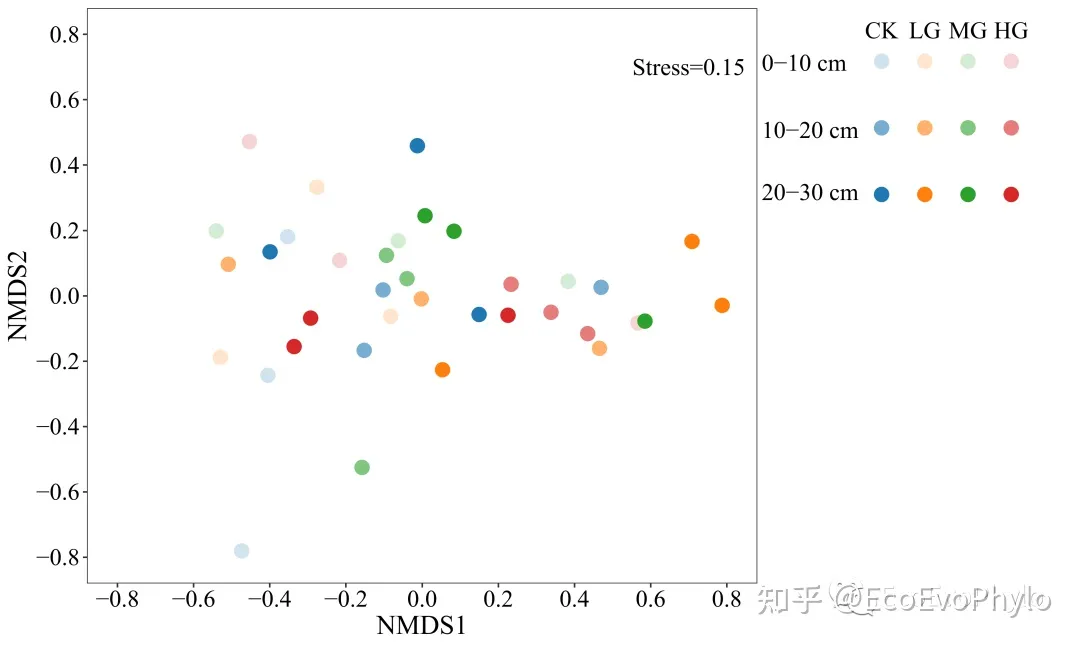
图10|NMDS样本散点图,p1.pdf。
颜色超过三种,用渐变色区分分类因子,就不太直观了。所以下面使用不同颜色区分grazing,不同形状区分depth。
# 3.3 自定义图例-颜色区分grazing,形状区分depth。
## 3.3.1 颜色
library(ggthemes) # 更多主题
cols <- mypal[9:12]
show_col(cols)
## 3.3.2 绘图
p2 <- ggplot(data=mydata1,aes(MDS1,MDS2))+
geom_point(data=mydata1,aes(color=Condition,shape=depth),
size=4.5,alpha=0.7)+
#stat_ellipse(aes(group=grazing,color=grazing),level = 0.95,linetype=2)+ # 此句可添加置信圈,此函数不能为只有三个点的组添加置信圈。只有三个点的可使用ggforce包添加置信圈。
scale_color_manual(values = rep(cols,times=4),
breaks = c( # 自定义分类变量顺序,与颜色顺序对应。
"CK-A","LG-A","MG-A","HG-A",
"CK-B","LG-B","MG-B","HG-B",
"CK-C","LG-C","MG-C","HG-C"
),
labels = c(
"CK","LG","MG","HG",
rep("",8)
))+
scale_shape_manual(values = c(16,17,15),guide="none")+ # ?points查看数字对应点形状
theme_base()+
labs(x="NMDS1","NMDS2")+
annotate("text",x=0.7,y=0.7,size=7,colour = "black",# x,y设置stress值的放置位置。
label="paste('Stress=',0.15)",parse = TRUE)+
guides(color =guide_legend(override.aes = list(shape =rep(c(16,17,15),each=4)),
title=paste("0-10 cm","10-20 cm","20-30 cm",
sep = "\n\n\n"),
title.hjust = 1.5,
title.vjust = 1,
label.position = "left",
label.hjust =0,
keyheight = 2,
keywidth = 1.8,
ncol=3,
byrow = FALSE))+ # 设置图例,参数意义可查看函数帮助信息。
theme(
axis.title = element_text(size=16,color = "black"),
axis.text = element_text(size=14,color = "black"),
legend.text = element_text(size=14,color="black"),
legend.title = element_text(size=14,colour = "black",angle = 90),
)
p2
## 3.3.3 输出图到本地
pdf("p2.pdf",height = 6,width = 10,family = "Times")
print(p2)#输出图的depth需自己对齐一下。
dev.off()
图11|NMDS样本散点图,p2.pdf。
四、多元统计分析
帮助文档中提示分析组间多元变量的差异,ANOSIM与NMDS一般搭配使用。但此数据有两个分类变量(存在嵌套),选择adonis可以分析两个分类的交互效应。
# 4.1 Permutational Multivariate Analysis of Variance Using Distance Matrices
set.seed(12345)
t1 <- adonis2(spe[-c(1:3)] ~ grazing*depth, data = spe,
permutations = 999, method = "bray",
by = "terms") # 输出所有terms的检验结果,会受因子顺序影响。
##注意区分grazing+depth,grazing*depth和grazing:depth。
t2 <- adonis2(spe[-c(1:3)] ~ grazing+depth, data = spe,
permutations = 999, method = "bray",
by = "margin") # 输出边际效应
t3 <- adonis2(spe[-c(1:3)] ~ grazing*depth, data = spe,
permutations = 999, method = "bray",
by = NULL) # 整个模型的检验结果
t1
t2
t3
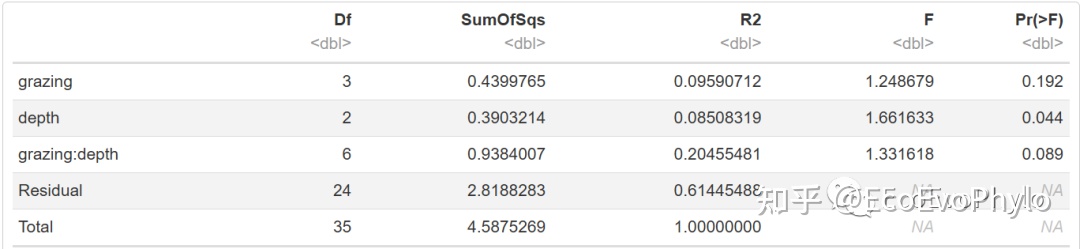
图12|adonis2多元统计结果,t1。
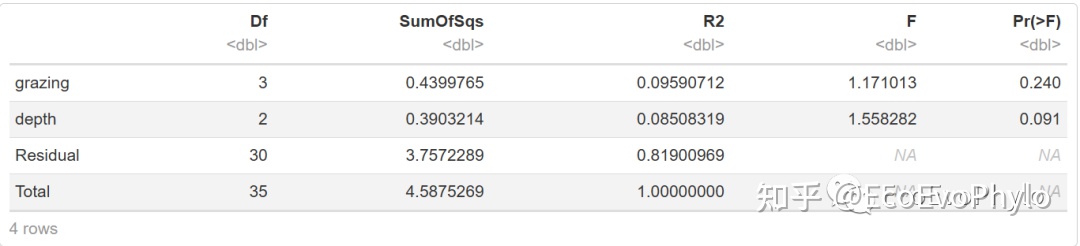
图13|adonis2多元统计结果,t2。

图14|adonis2多元统计结果,t3。
# 4.2 限制分层置换
set.seed(12345)
t4 <- adonis2(spe[-c(1:3)] ~ grazing, data = spe, permutations =999, strata = spe$depth) # 限制depth置换,检验grazing效应
t5 <- adonis2(spe[-c(1:3)] ~ depth, data = spe, permutations = 999, strata = spe$grazing) # 限制grazing置换,检验depth效应
t4
t5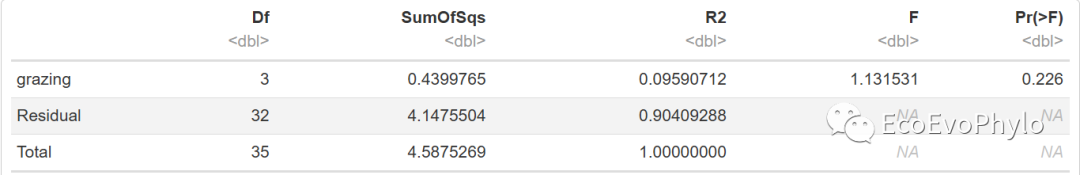
图15|adonis2多元统计结果,t4。
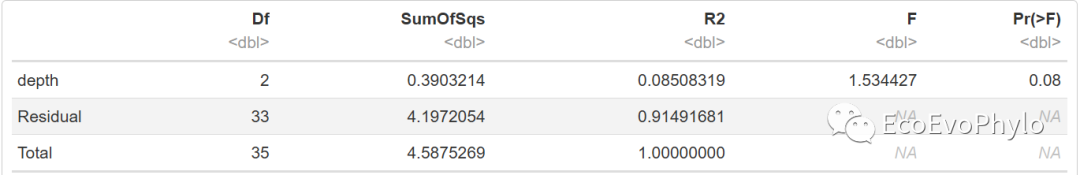
图16|adonis2多元统计结果,t5。
# 4.3 ANOSIM群落结构差异分析-bray index
set.seed(12345)
grazing.anosim <- anosim(spe[-c(1:3)],
spe$grazing,
permutations = 999,distance = "bray")
depth.anosim <- anosim(spe[-c(1:3)],
spe$depth,
permutations = 999,distance = "bray")
grazing.anosim
depth.anosim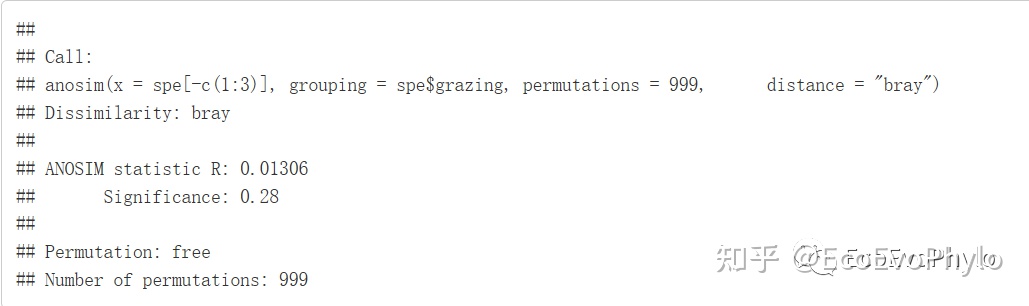
图17|ANOSIM多元统计结果,grazing.anosim。

图18|ANOSIM多元统计结果,depth.anosim。
更多结果解读及绘图,参考R统计-微生物群落结构差异分析及结果解读。
五、拟合环境变量
5.1 线性拟合-envfit()
线性拟合只能拟合物种与环境因子的线性关系。
# 5.1.1 相关性系数
env.cor <- cor(env[-c(1:3)])
env.cor # 选择相关性较低和感兴趣的2个变量作为示例。
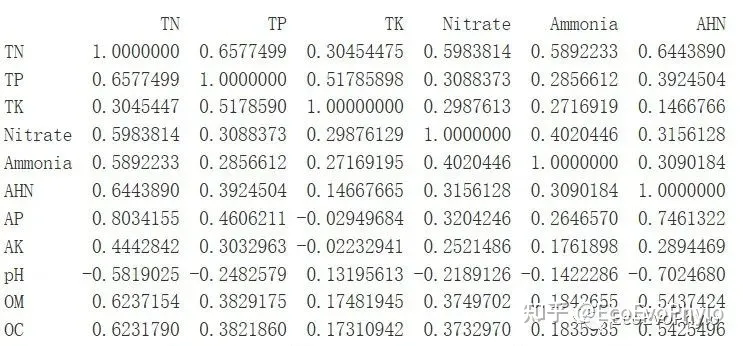
图19|环境因子相关系数,env.cor。
# 5.1.2 envfit()线性拟合环境因子
##选择相关性较低和感兴趣的2个变量作为示例。
set.seed(12345)
NMDS1.env <- envfit(NMDS1 ~ TN+TK, env)
NMDS1.env
name <- names(which(apply(spe[-c(1:3)],2,mean) > 0.01)) # otu太多,只展示平均相对丰度大于0.01的otu。

图20|NMDS1环境因子拟合结果,NMDS1.env。
# 5.1.3 绘图
pdf("envfit.pdf",width = 6,height = 8,family = "Times")
plot(NMDS1,display = "sites",
xlim = c(-0.8,1), ylim=c(-1,0.8),
main = "NMDS species relative abundance",
sub = "Fitted linear: TN + TK")
abline(h=0,lty=2);abline(v=0,lty=2)
text(NMDS1$points,rownames(spe),cex=(1-goodness(NMDS1)),col = "black") #添加样本标签,字体越大,排序结果代表性越好。
text(NMDS1$species[name,],name,cex=0.5,col="red")
plot(NMDS1.env,lwd=1) # 3 arrows
dev.off()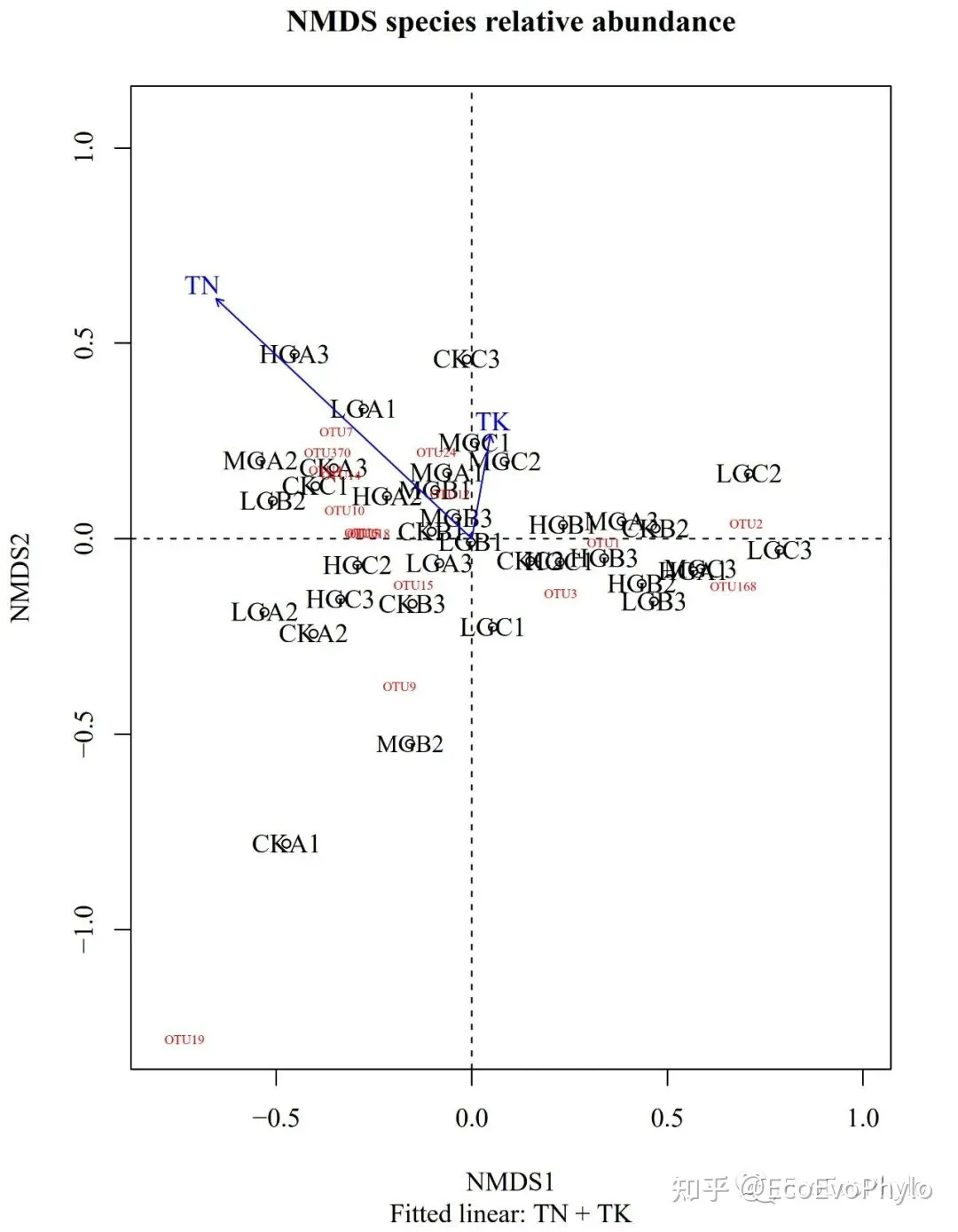
图21|NMDS1环境因子线性拟合图,envfit.pdf。
5.2 曲面拟合-ordisurf()或surf()
环境因子曲面拟合可以在非线性基础上,展示环境因子与排序结果的关联。ordisurf()和surf()使用的是广义相加模型(GAM)。
# 5.2.1 vegan::ordisurf()拟合环境因子到NMDS1排序图中
pdf("ordisurf.pdf",width = 6,height = 8,family = "Times")
set.seed(12345)
plot(NMDS1,display = "sites", type = "n",
#xlim = c(-0.8,1), ylim=c(-1,0.8),
main = "NMDS species relative abundance",
sub = "Fitted curve: TN(DarkKhaki) + TK(Tomato)")
abline(h=0,lty=2);abline(v=0,lty=2)
points(NMDS1,display = "sites",
lwd = 1,
col = rep(cols,each=3),# 按grazing设置点颜色。
pch = rep(c(16,17,15),each=12))# 按depth设置点形状。
#text(NMDS1$points,rownames(spe),cex= 0.8,col = "black")# 不展示样本标签
text(NMDS1$species[name,],name,cex=0.5,col="black")
plot(NMDS1.env,lwd=1,col = c("blue")) # 3 arrows
ordisurf(NMDS1~TN, env,add=TRUE,col="#BDB76B",
family = "gaussian") # 更多参数设置?ordisurf查看
ordisurf(NMDS1~TK, env,add=TRUE,col="#FF6347", family = "gaussian")
legend(x = 0.1,y=-1,legend = unique(spe$Condition),
col = cols,pch = rep(c(16,17,15),each=4),
xjust = 0,yjust = 0,# 以左下为基准对齐
ncol = 3)
dev.off()
## vegan包中还有很多强大的函数,可以看帮助文件进行学习。
tabasco(spe[-c(1:3)],NMDS1) # 将物种按照NMDS结果进行排序并绘制热图。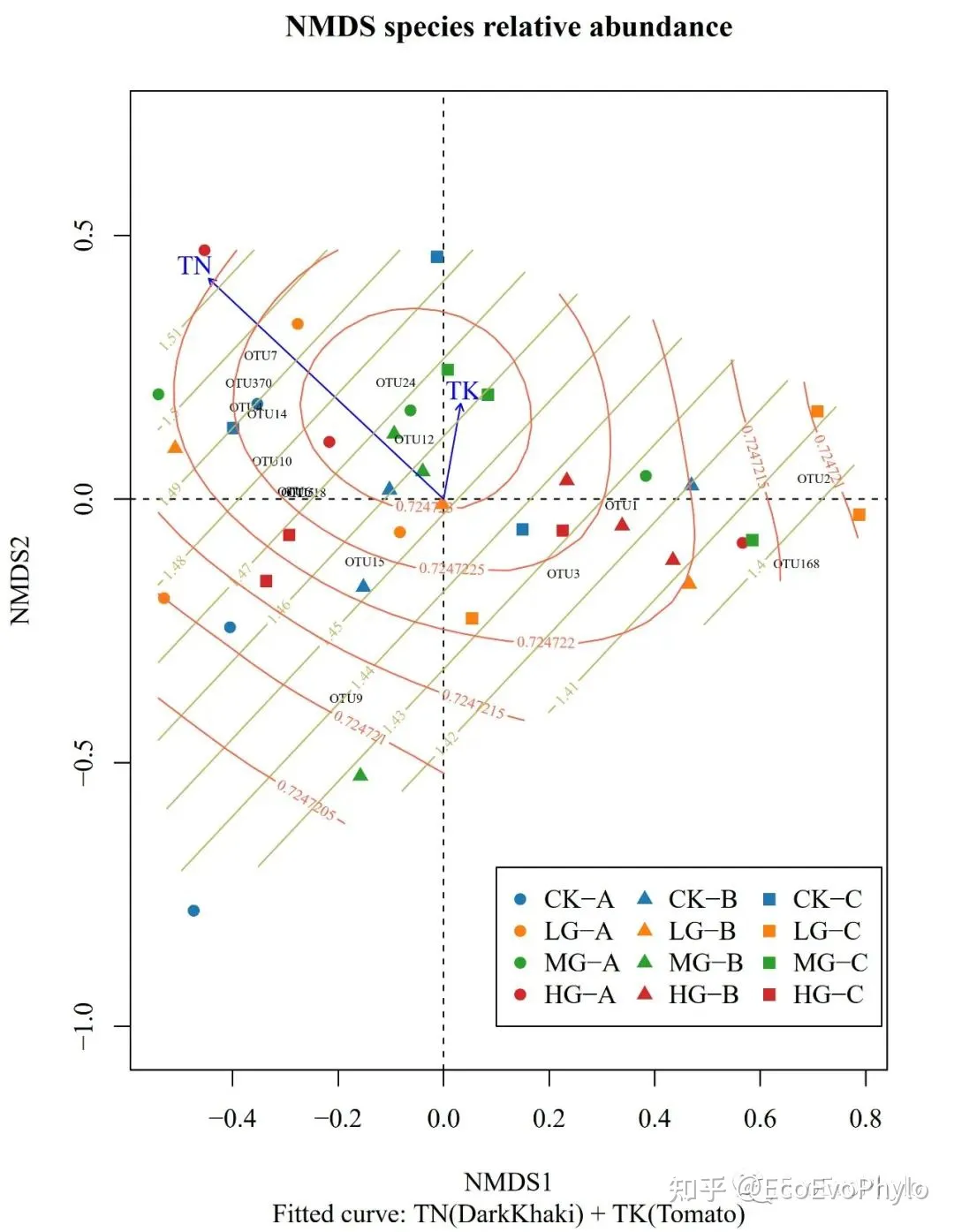
图22|NMDS1环境因子曲面(GAM)拟合图,ordisurf.pdf。由图可以看出TK与NMDS排序关系是非线性的,而TN是线性的(曲面由平行直线组成)。曲面上标记的是环境因子的数值。
## 5.2.2 labdsv::surf()拟合环境因子到NMDS2排序图中
pdf("surf.pdf",width = 6,height = 8,family = "Times")
set.seed(12345)
plot(NMDS2,col="white")
points(NMDS2$points,
lwd = 1,
col = rep(cols,each=3),# 按grazing设置点颜色。
pch = rep(c(16,17,15),each=12))# 按depth设置点形状。
#text(NMDS2$points,rownames(spe))
text(species2[name,],name,cex=0.5,col="black")
surf(NMDS2,env$TN,col="#BDB76B",thinplate=FALSE)
surf(NMDS2,env$TK,col="#FF6347",thinplate=FALSE)
legend(x = -15,y=-20,legend = unique(spe$Condition),
col = cols,pch = rep(c(16,17,15),each=4),
xjust = 0,yjust = 0,# 以左下为基准对齐
ncol = 3)
dev.off()
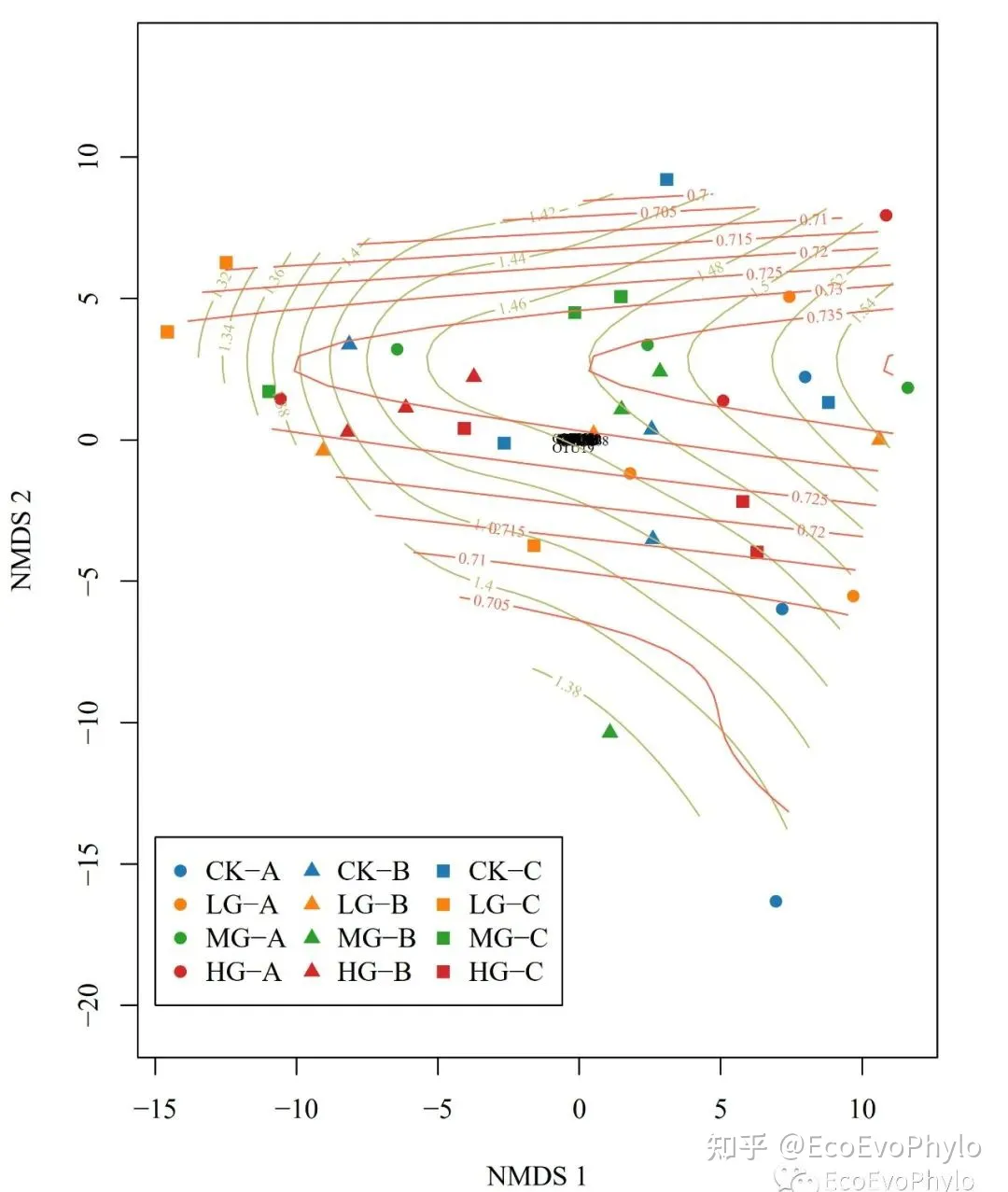
图23|NMDS2环境因子曲面(GAM)拟合图,surf.pdf。番茄色为TK,深卡其布色为TN。此拟合结果显示TK和TN与NMDS2都非线性关系。
两个函数计算出的样本得分、物种得分和环境因子拟合结果差异有一些大。NMDS由于是随机启动的,容易产生次优解。所以NMDS最好进行多次运行。因为涉及随机过程,为保证结果的可重复性,最好运行时设置随机种子。个人觉得还是基于特征分解的PCoA的分析结果更稳定,不受随机的影响,且结果更易解释,《Numerical Ecology with R》中也更推荐PCoA。书中提到PCoA等的排序结果,可以作为NMDS的输入,用于将超过2-3D的数据空间,在2-3D的维度展示出来。
数据表和代码可从QQ交流群文件夹中下载,或EcoEvoPhylo公众号后台发送“NMDS详解1”获取。
原文链接:R统计绘图-NMDS、环境因子拟合(线性和非线性)、多元统计(adonis2和ANOSIM)及绘图(双因素自定义图例) (qq.com)
参考资料
手动添加图例:请问ggplot2如何给自己的图增加图例? - 知乎图例设置:https://blog.csdn.net/tanzuozhev/article/details/51108040[NMDS分析](NMDS分析 - 知乎)[生信学习-高通量分析-NMDS分析: 非度量多维尺度分析](https://blog.csdn.net/weixin_53819139/article/details/114133818)R统计-微生物群落结构差异分析及结果解读
R统计-PCA/PCoA/db-RDA/NMDS/CA/CCA/DCA等排序分析教程Borcard, Daniel, François Gillet and Pierre Legendre. Numerical Ecology with R. Use R! Cham: Springer International Publishing, 2018. https://doi.org/10.1007/978-3-319-71404-2.
推荐阅读
R绘图-物种、环境因子相关性网络图(简单图、提取子图、修改图布局参数、物种-环境因子分别成环径向网络图)
R统计绘图-分子生态相关性网络分析(拓扑属性计算,ggraph绘图)
R统计绘图-变量分组相关性网络图(igraph)
机器学习-分类随机森林分析(randomForest模型构建、参数调优、特征变量筛选、模型评估和基础理论等)
R统计绘图-环境因子相关性+mantel检验组合图(linkET包介绍1)
R统计绘图-NMDS、环境因子拟合(线性和非线性)、多元统计(adonis2和ANOSIM)及绘图(双因素自定义图例)
R统计绘图-PCA详解1(princomp/principal/rcomp/rda等)
R统计-PCA/PCoA/db-RDA/NMDS/CA/CCA/DCA等排序分析教程
R统计绘图-PCA分析绘图及结果解读(误差线,多边形,双Y轴图、球形检验、KMO和变量筛选等)
R统计绘图-corrplot热图绘制细节调整2(更改变量可视化顺序、非相关性热图绘制、添加矩形框等)
R统计绘图 | 物种组成冲积图(绝对/相对丰度,ggalluvial)
R统计绘图 | 物种组成堆叠面积图(绝对/相对丰度,ggalluvial)
R统计-单因素ANOVA/Kruskal-Wallis置换检验
R统计绘图-单、双、三因素重复测量方差分析[Translation]



















 1092
1092

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











