








点击关注,桓峰基因
桓峰基因
生物信息分析,SCI文章撰写及生物信息基础知识学习:R语言学习,perl基础编程,linux系统命令,Python遇见更好的你
81篇原创内容
公众号
转发朋友圈获得更多培训资料和代码,扫码加微信,成为顶级生信工程师,实现年薪30W+!

前 言
随着癌症基因组学的进步,突变注释格式(MAF)正在被广泛接受并用于存储检测到的体细胞变异。癌症基因组图谱项目已经对30多种不同的癌症进行了测序,每种癌症的样本量都超过了200个。由体细胞变体组成的结果数据以突变注释格式的形式存储。只要数据是MAF格式,本包试图从TCGA来源或任何内部研究中以有效的方式总结、分析、注释和可视化MAF文件。
实例解析
1. 软件安装
在安装这个软件maftools时,需要先安装BioManager,然后在安装maftools,如下:
if (!require("BiocManager")) {
install.packages("BiocManager")
}
if (!require(maftools)) {
BiocManager::install("maftools")
}
library(maftools)
2. 数据读取
maftools工具需要读入两个文件,如下:
1.MAF文件-可以是gz压缩。必需的;
2.与MAF中每个样本/肿瘤样本条码相关的可选但推荐的临床数据;
3.一个可选的拷贝数数据:可以是GISTIC输出或自定义表。
1.maf文件格式
MAF文件包含许多字段,从染色体名称到cosmic注释。然而,maftools中的大多数分析使用以下列如下:1.Hugo_Symbol;
2.Chromosome;
3.Start_Position;
4.End_Position;
5.Reference_Allele;
6.Tumor_Seq_Allele2;
7.Variant_Classification;
8.Variant_Type;
9.Tumor_Sample_Barcode.
同时读取maf文件和临床信息文件,看下结果,如下:
# path to TCGA LAML MAF file
laml.maf = system.file("extdata", "tcga_laml.maf.gz", package = "maftools")
# clinical information containing survival information and histology. This is
# optional
laml.clin = system.file("extdata", "tcga_laml_annot.tsv", package = "maftools")
laml = read.maf(maf = laml.maf, clinicalData = laml.clin)
## -Reading
## -Validating
## -Silent variants: 475
## -Summarizing
## -Processing clinical data
## -Finished in 3.520s elapsed (0.690s cpu)
print(laml@data[1:5, ])
## Hugo_Symbol Entrez_Gene_Id Center NCBI_Build Chromosome
## 1: ABCA10 10349 genome.wustl.edu 37 17
## 2: ABCA4 24 genome.wustl.edu 37 1
## 3: ABCB11 8647 genome.wustl.edu 37 2
## 4: ABCC3 8714 genome.wustl.edu 37 17
## 5: ABCF1 23 genome.wustl.edu 37 6
## Start_Position End_Position Strand Variant_Classification Variant_Type
## 1: 67170917 67170917 + Splice_Site SNP
## 2: 94490594 94490594 + Missense_Mutation SNP
## 3: 169780250 169780250 + Missense_Mutation SNP
## 4: 48760974 48760974 + Missense_Mutation SNP
## 5: 30554429 30554429 + Missense_Mutation SNP
## Reference_Allele Tumor_Seq_Allele1 Tumor_Seq_Allele2 Tumor_Sample_Barcode
## 1: T T C TCGA-AB-2988
## 2: C C T TCGA-AB-2869
## 3: G G A TCGA-AB-3009
## 4: C C T TCGA-AB-2887
## 5: G G A TCGA-AB-2920
## Protein_Change i_TumorVAF_WU i_transcript_name
## 1: p.K960R 45.66000 NM_080282.3
## 2: p.R1517H 38.12000 NM_000350.2
## 3: p.A1283V 46.97218 NM_003742.2
## 4: p.P1271S 56.41000 NM_003786.1
## 5: p.G658S 40.95000 NM_001025091.1
2.临床信息格式
临床数据格式包括:每个样本/肿瘤样本条码,和对应的临床数据,如下:
print(laml@clinical.data[1:5, ])
## Tumor_Sample_Barcode FAB_classification days_to_last_followup
## 1: TCGA-AB-2802 M4 365
## 2: TCGA-AB-2803 M3 792
## 3: TCGA-AB-2804 M3 2557
## 4: TCGA-AB-2805 M0 577
## 5: TCGA-AB-2806 M1 945
## Overall_Survival_Status
## 1: 1
## 2: 1
## 3: 0
## 4: 1
## 5: 1
3.拷贝数变异格式
拷贝数数据包含样本名称,基因名称和拷贝数状态(Amp或Del)。
all.lesions <- system.file("extdata", "all_lesions.conf_99.txt", package = "maftools")
amp.genes <- system.file("extdata", "amp_genes.conf_99.txt", package = "maftools")
del.genes <- system.file("extdata", "del_genes.conf_99.txt", package = "maftools")
scores.gis <- system.file("extdata", "scores.gistic", package = "maftools")
laml.gistic = readGistic(gisticAllLesionsFile = all.lesions, gisticAmpGenesFile = amp.genes,
gisticDelGenesFile = del.genes, gisticScoresFile = scores.gis, isTCGA = TRUE)
## -Processing Gistic files..
## --Processing amp_genes.conf_99.txt
## --Processing del_genes.conf_99.txt
## --Processing scores.gistic
## --Summarizing by samples
laml.gistic@data[1:5, ]
## Hugo_Symbol Tumor_Sample_Barcode Variant_Classification Variant_Type
## 1: KMT2A TCGA-AB-2805 Amp CNV
## 2: LINC00689 TCGA-AB-2805 Del CNV
## 3: MIR595 TCGA-AB-2805 Del CNV
## 4: RN7SL142P TCGA-AB-2805 Del CNV
## 5: SHH TCGA-AB-2805 Del CNV
## Cytoband
## 1: AP_2:11q23.3
## 2: DP_4:7q32.3
## 3: DP_4:7q32.3
## 4: DP_4:7q32.3
## 5: DP_4:7q32.3
4.突变分析
突变基因的互作
相互排斥或同时出现的基因集可以通过somaticInteractions功能进行检测,该功能执行成对的Fisher-test,以检测这样的重要基因对。
# exclusive/co-occurance event analysis on top 10 mutated genes.
somaticInteractions(maf = laml, top = 25, pvalue = c(0.05, 0.1))

## gene1 gene2 pValue oddsRatio 00 11 01 10 Event
## 1: ASXL1 RUNX1 0.0001541586 55.215541 176 4 12 1 Co_Occurence
## 2: IDH2 RUNX1 0.0002809928 9.590877 164 7 9 13 Co_Occurence
## 3: IDH2 ASXL1 0.0004030636 41.077327 172 4 1 16 Co_Occurence
## 4: FLT3 NPM1 0.0009929836 3.763161 125 17 16 35 Co_Occurence
## 5: SMC3 DNMT3A 0.0010451985 20.177713 144 6 42 1 Co_Occurence
## ---
## 296: PLCE1 ASXL1 1.0000000000 0.000000 184 0 5 4 Mutually_Exclusive
## 297: RAD21 FAM5C 1.0000000000 0.000000 183 0 5 5 Mutually_Exclusive
## 298: PLCE1 FAM5C 1.0000000000 0.000000 184 0 5 4 Mutually_Exclusive
## 299: PLCE1 RAD21 1.0000000000 0.000000 184 0 5 4 Mutually_Exclusive
## 300: EZH2 PLCE1 1.0000000000 0.000000 186 0 4 3 Mutually_Exclusive
## pair event_ratio
## 1: ASXL1, RUNX1 4/13
## 2: IDH2, RUNX1 7/22
## 3: ASXL1, IDH2 4/17
## 4: FLT3, NPM1 17/51
## 5: DNMT3A, SMC3 6/43
## ---
## 296: ASXL1, PLCE1 0/9
## 297: FAM5C, RAD21 0/10
## 298: FAM5C, PLCE1 0/9
## 299: PLCE1, RAD21 0/9
## 300: EZH2, PLCE1 0/7
基于位置聚类的癌症驱动基因检测
maftools有一个功能癌基因驱动器,可以从一个给定的MAF中识别癌症基因(驱动器)。oncodrive是一个基于算法oncodriveCLUST的软件,它最初是用Python实现的。概念是基于这样的事实,即大多数变异的致癌基因在少数特定位点(又名热点)富集。这种方法利用这些位置来识别癌症基因。
laml.sig = oncodrive(maf = laml, AACol = "Protein_Change", minMut = 5, pvalMethod = "zscore")
head(laml.sig)
## Hugo_Symbol Frame_Shift_Del Frame_Shift_Ins In_Frame_Del In_Frame_Ins
## 1: IDH1 0 0 0 0
## 2: IDH2 0 0 0 0
## 3: NPM1 0 33 0 0
## 4: NRAS 0 0 0 0
## 5: U2AF1 0 0 0 0
## 6: KIT 1 1 0 1
## Missense_Mutation Nonsense_Mutation Splice_Site total MutatedSamples
## 1: 18 0 0 18 18
## 2: 20 0 0 20 20
## 3: 1 0 0 34 33
## 4: 15 0 0 15 15
## 5: 8 0 0 8 8
## 6: 7 0 0 10 8
## AlteredSamples clusters muts_in_clusters clusterScores protLen zscore
## 1: 18 1 18 1.0000000 414 5.546154
## 2: 20 2 20 1.0000000 452 5.546154
## 3: 33 2 32 0.9411765 294 5.093665
## 4: 15 2 15 0.9218951 189 4.945347
## 5: 8 1 7 0.8750000 240 4.584615
## 6: 8 2 9 0.8500000 976 4.392308
## pval fdr fract_muts_in_clusters
## 1: 1.460110e-08 1.022077e-07 1.0000000
## 2: 1.460110e-08 1.022077e-07 1.0000000
## 3: 1.756034e-07 8.194826e-07 0.9411765
## 4: 3.800413e-07 1.330144e-06 1.0000000
## 5: 2.274114e-06 6.367520e-06 0.8750000
## 6: 5.607691e-06 1.308461e-05 0.9000000
plotOncodrive(res = laml.sig, fdrCutOff = 0.1, useFraction = TRUE, labelSize = 0.5)
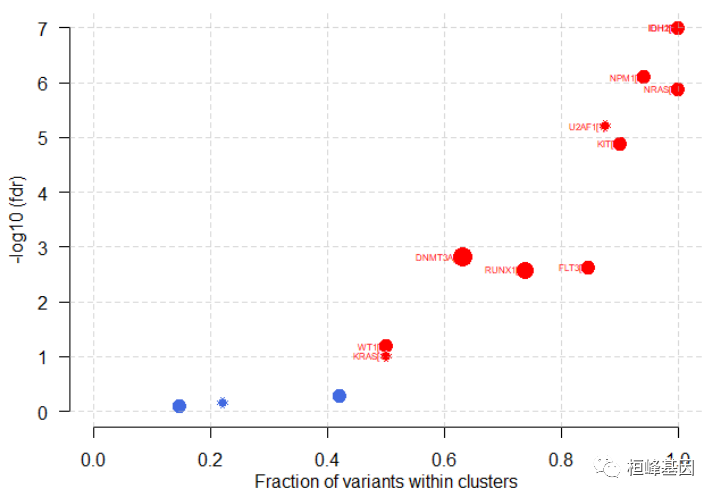
添加和总结 pfam domains
maftools具有pfamDomains功能,它将pfam域信息添加到氨基酸变化中。pfamDomain还根据受影响的域总结了氨基酸的变化。这有助于了解在给定的癌症队列中,哪些领域最常受影响。
laml.pfam = pfamDomains(maf = laml, AACol = "Protein_Change", top = 10)
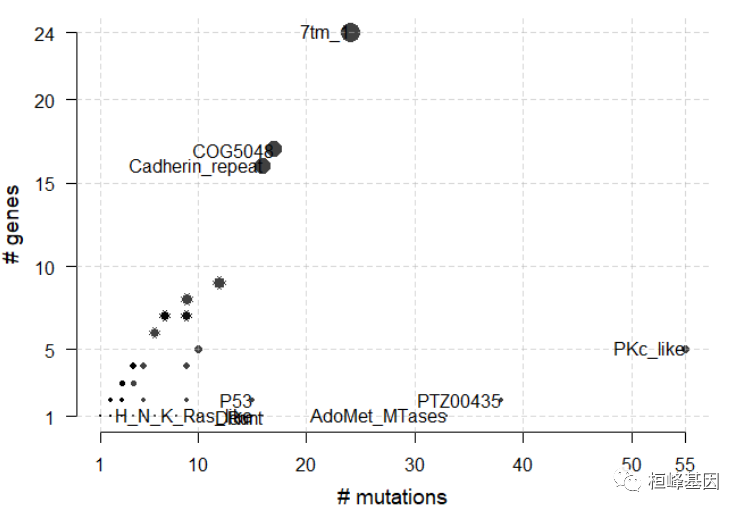
# Protein summary (Printing first 7 columns for display convenience)
laml.pfam$proteinSummary[, 1:7, with = FALSE]
## HGNC AAPos Variant_Classification N total fraction DomainLabel
## 1: DNMT3A 882 Missense_Mutation 27 54 0.5000000 AdoMet_MTases
## 2: IDH1 132 Missense_Mutation 18 18 1.0000000 PTZ00435
## 3: IDH2 140 Missense_Mutation 17 20 0.8500000 PTZ00435
## 4: FLT3 835 Missense_Mutation 14 52 0.2692308 PKc_like
## 5: FLT3 599 In_Frame_Ins 10 52 0.1923077 PKc_like
## ---
## 1512: ZNF646 875 Missense_Mutation 1 1 1.0000000 <NA>
## 1513: ZNF687 554 Missense_Mutation 1 2 0.5000000 <NA>
## 1514: ZNF687 363 Missense_Mutation 1 2 0.5000000 <NA>
## 1515: ZNF75D 5 Missense_Mutation 1 1 1.0000000 <NA>
## 1516: ZNF827 427 Frame_Shift_Del 1 1 1.0000000 <NA>
laml.pfam$domainSummary[, 1:3, with = FALSE]
## DomainLabel nMuts nGenes
## 1: PKc_like 55 5
## 2: PTZ00435 38 2
## 3: AdoMet_MTases 33 1
## 4: 7tm_1 24 24
## 5: COG5048 17 17
## ---
## 499: ribokinase 1 1
## 500: rim_protein 1 1
## 501: sigpep_I_bact 1 1
## 502: trp 1 1
## 503: zf-BED 1 1
生存分析
生存分析是队列测序项目的重要组成部分。mafsurvival功能mafsurvival进行生存分析并绘制kaplan meier曲线,根据用户定义的基因的突变状态对样本进行分组,或者手工提供组成一个组的样本。该函数要求输入数据包含Tumor_Sample_Barcode(确保它们与MAF文件中的匹配)、二进制事件(1/0)和事件发生时间。我们的注释数据已经包含生存信息,如果您有生存数据存储在单独的表中,请通过参数clinicalData提供它们。
- 指定突变基因
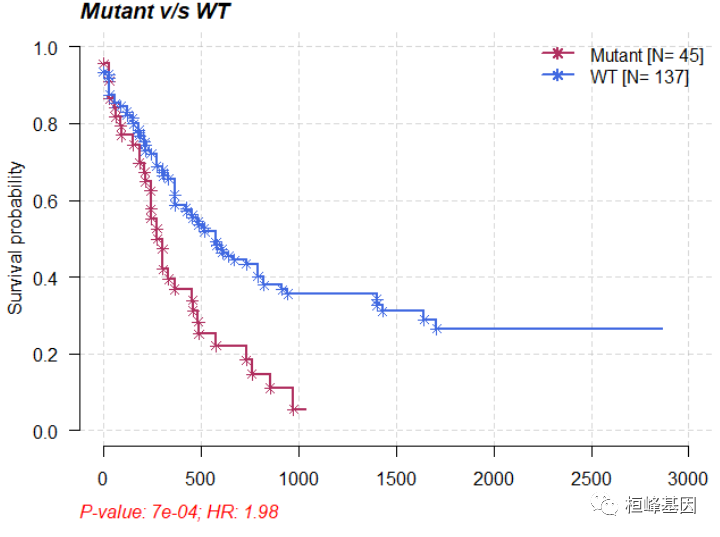
# Survival analysis based on grouping of DNMT3A mutation status
mafSurvival(maf = laml, genes = "DNMT3A", time = "days_to_last_followup", Status = "Overall_Survival_Status",
isTCGA = TRUE)
## DNMT3A
## 48
## Group medianTime N
## 1: Mutant 245 45
## 2: WT 396 137
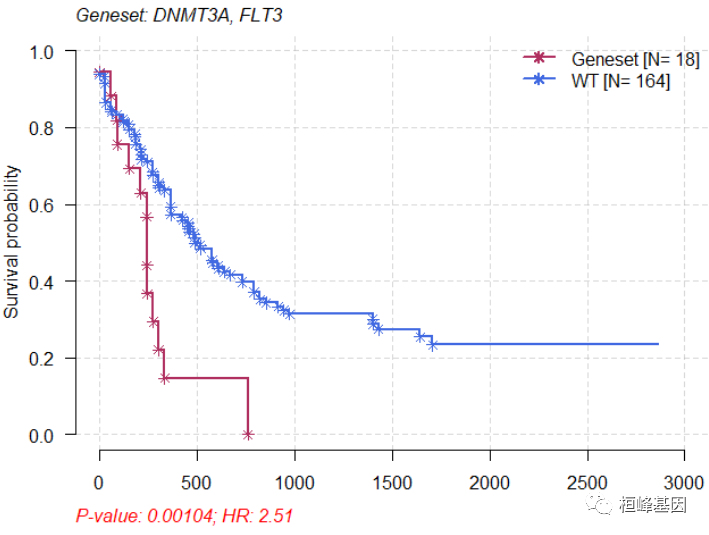
- 相关基因集
# Using top 20 mutated genes to identify a set of genes (of size 2) to predict
# poor prognostic groups
prog_geneset = survGroup(maf = laml, top = 20, geneSetSize = 2, time = "days_to_last_followup",
Status = "Overall_Survival_Status", verbose = FALSE)
print(prog_geneset)
## Gene_combination P_value hr WT Mutant
## 1: FLT3_DNMT3A 0.00104 2.510 164 18
## 2: DNMT3A_SMC3 0.04880 2.220 176 6
## 3: DNMT3A_NPM1 0.07190 1.720 166 16
## 4: DNMT3A_TET2 0.19600 1.780 176 6
## 5: FLT3_TET2 0.20700 1.860 177 5
## 6: NPM1_IDH1 0.21900 0.495 176 6
## 7: DNMT3A_IDH1 0.29300 1.500 173 9
## 8: IDH2_RUNX1 0.31800 1.580 176 6
## 9: FLT3_NPM1 0.53600 1.210 165 17
## 10: DNMT3A_IDH2 0.68000 0.747 178 4
## 11: DNMT3A_NRAS 0.99200 0.986 178 4
mafSurvGroup(maf = laml, geneSet = c("DNMT3A", "FLT3"), time = "days_to_last_followup",
Status = "Overall_Survival_Status")
## Group medianTime N
## 1: Mutant 242.5 18
## 2: WT 379.5 164
比较两个cohort
我们经常都会比较一个突变在原发和复发/转移的癌组织中的突变情况,简单的比较方法,如下:
# Primary APL MAF
primary.apl = system.file("extdata", "APL_primary.maf.gz", package = "maftools")
primary.apl = read.maf(maf = primary.apl)
## -Reading
## -Validating
## --Non MAF specific values in Variant_Classification column:
## ITD
## -Silent variants: 45
## -Summarizing
## -Processing clinical data
## --Missing clinical data
## -Finished in 4.920s elapsed (0.640s cpu)
# Relapse APL MAF
relapse.apl = system.file("extdata", "APL_relapse.maf.gz", package = "maftools")
relapse.apl = read.maf(maf = relapse.apl)
## -Reading
## -Validating
## --Non MAF specific values in Variant_Classification column:
## ITD
## -Silent variants: 19
## -Summarizing
## -Processing clinical data
## --Missing clinical data
## -Finished in 5.090s elapsed (0.680s cpu)
# Considering only genes which are mutated in at-least in 5 samples in one of
# the cohort to avoid bias due to genes mutated in single sample.
pt.vs.rt <- mafCompare(m1 = primary.apl, m2 = relapse.apl, m1Name = "Primary", m2Name = "Relapse",
minMut = 5)
print(pt.vs.rt)
## $results
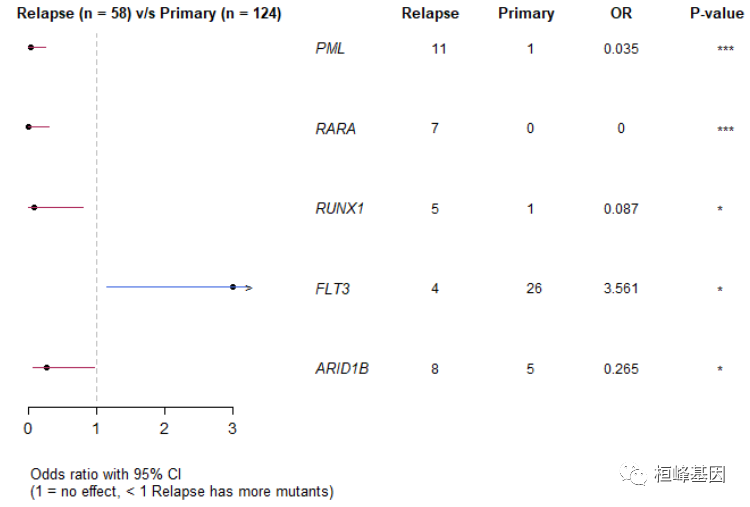
## Hugo_Symbol Primary Relapse pval or ci.up ci.low
## 1: PML 1 11 1.529935e-05 0.03537381 0.2552937 0.000806034
## 2: RARA 0 7 2.574810e-04 0.00000000 0.3006159 0.000000000
## 3: RUNX1 1 5 1.310500e-02 0.08740567 0.8076265 0.001813280
## 4: FLT3 26 4 1.812779e-02 3.56086275 14.7701728 1.149009169
## 5: ARID1B 5 8 2.758396e-02 0.26480490 0.9698686 0.064804160
## 6: WT1 20 14 2.229087e-01 0.60619329 1.4223101 0.263440988
## 7: KRAS 6 1 4.334067e-01 2.88486293 135.5393108 0.337679367
## 8: NRAS 15 4 4.353567e-01 1.85209500 8.0373994 0.553883512
## 9: ARID1A 7 4 7.457274e-01 0.80869223 3.9297309 0.195710173
## adjPval
## 1: 0.0001376942
## 2: 0.0011586643
## 3: 0.0393149868
## 4: 0.0407875250
## 5: 0.0496511201
## 6: 0.3343630535
## 7: 0.4897762916
## 8: 0.4897762916
## 9: 0.7457273717
##
## $SampleSummary
## Cohort SampleSize
## 1: Primary 124
## 2: Relapse 58
- 森林图 我们可以通过fisher检验,可视化每个突变在不同时期的差异,如下:
forestPlot(mafCompareRes = pt.vs.rt, pVal = 0.1)
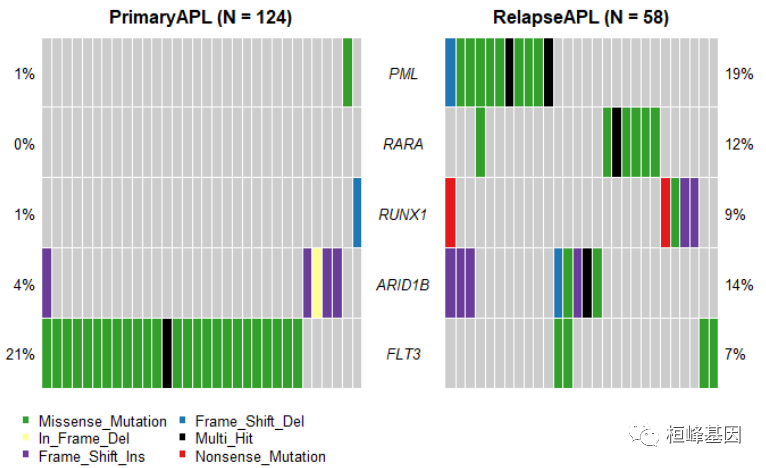
- 分组瀑布图 通过fisher检验得到差异突变基因,然后绘制两个分组的瀑布图,如下:
genes = c("PML", "RARA", "RUNX1", "ARID1B", "FLT3")
coOncoplot(m1 = primary.apl, m2 = relapse.apl, m1Name = "PrimaryAPL", m2Name = "RelapseAPL",
genes = genes, removeNonMutated = TRUE)
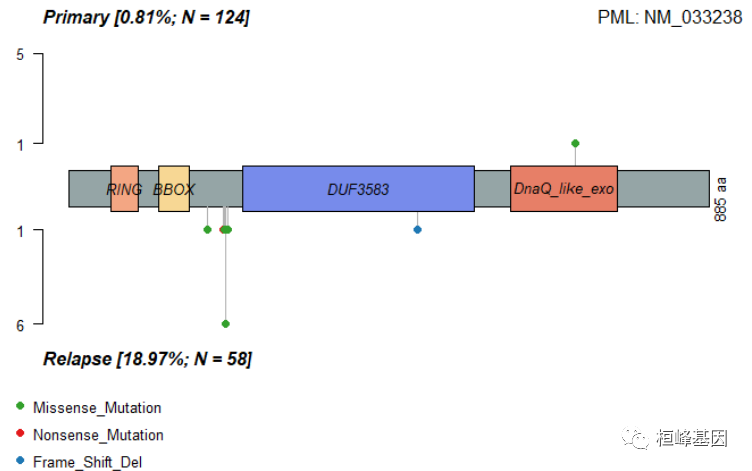
- 分组棒棒糖图 通过棒棒糖图来展示不同组的突变情况,如下:
lollipopPlot2(m1 = primary.apl, m2 = relapse.apl, gene = "PML", AACol1 = "amino_acid_change",
AACol2 = "amino_acid_change", m1_name = "Primary", m2_name = "Relapse")
## HGNC refseq.ID protein.ID aa.length
## 1: PML NM_002675 NP_002666 633
## 2: PML NM_033238 NP_150241 882
## 3: PML NM_033239 NP_150242 829
## 4: PML NM_033240 NP_150243 611
## 5: PML NM_033244 NP_150247 560
## 6: PML NM_033246 NP_150249 423
## 7: PML NM_033247 NP_150250 435
## 8: PML NM_033249 NP_150252 585
## 9: PML NM_033250 NP_150253 781
## HGNC refseq.ID protein.ID aa.length
## 1: PML NM_002675 NP_002666 633
## 2: PML NM_033238 NP_150241 882
## 3: PML NM_033239 NP_150242 829
## 4: PML NM_033240 NP_150243 611
## 5: PML NM_033244 NP_150247 560
## 6: PML NM_033246 NP_150249 423
## 7: PML NM_033247 NP_150250 435
## 8: PML NM_033249 NP_150252 585
## 9: PML NM_033250 NP_150253 781
临床富集分析
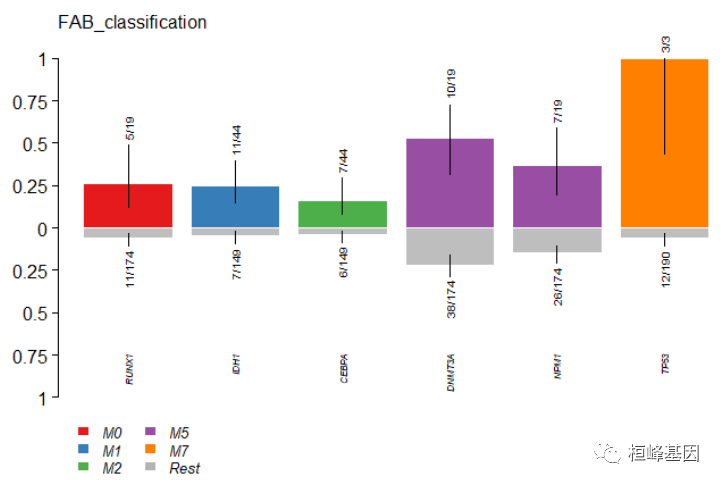
clinicalEnrichment是另一个功能,它取任何与样本相关的临床特征并进行富集分析。它执行各种分组和两两比较,以确定clinicila特征中每个类别的丰富突变。下面是一个识别与FAB_classification相关突变的示例。
fab.ce = clinicalEnrichment(maf = laml, clinicalFeature = "FAB_classification")
##
## M0 M1 M2 M3 M4 M5 M6 M7
## 19 44 44 21 39 19 3 3
# Results are returned as a list. Significant associations p-value < 0.05
fab.ce$groupwise_comparision[p_value < 0.05]
## Hugo_Symbol Group1 Group2 n_mutated_group1 n_mutated_group2 p_value
## 1: IDH1 M1 Rest 11 of 44 7 of 149 0.0002597371
## 2: TP53 M7 Rest 3 of 3 12 of 190 0.0003857187
## 3: DNMT3A M5 Rest 10 of 19 38 of 174 0.0089427384
## 4: CEBPA M2 Rest 7 of 44 6 of 149 0.0117352110
## 5: RUNX1 M0 Rest 5 of 19 11 of 174 0.0117436825
## 6: NPM1 M5 Rest 7 of 19 26 of 174 0.0248582372
## 7: NPM1 M3 Rest 0 of 21 33 of 172 0.0278630823
## 8: DNMT3A M3 Rest 1 of 21 47 of 172 0.0294005111
## OR OR_low OR_high fdr
## 1: 6.670592 2.173829026 21.9607250 0.0308575
## 2: Inf 5.355415451 Inf 0.0308575
## 3: 3.941207 1.333635173 11.8455979 0.3757978
## 4: 4.463237 1.204699322 17.1341278 0.3757978
## 5: 5.216902 1.243812880 19.4051505 0.3757978
## 6: 3.293201 1.001404899 10.1210509 0.5880102
## 7: 0.000000 0.000000000 0.8651972 0.5880102
## 8: 0.133827 0.003146708 0.8848897 0.5880102
plotEnrichmentResults(enrich_res = fab.ce, pVal = 0.05, geneFontSize = 0.5, annoFontSize = 0.6)
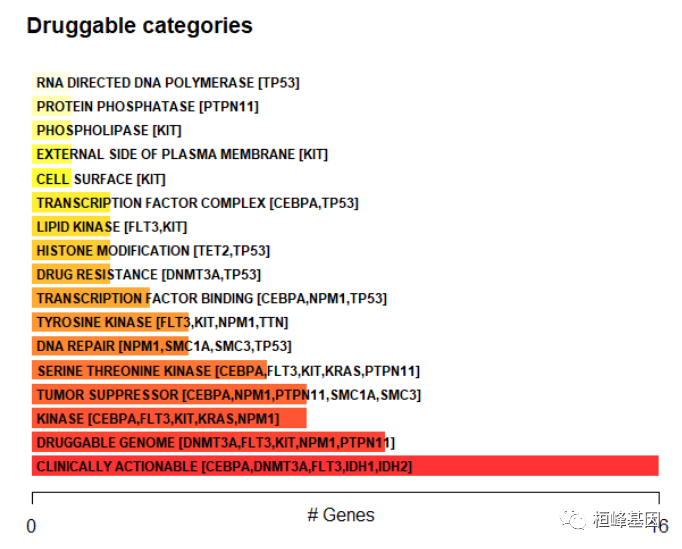
药物-基因互作
药物-基因相互作用功能检查和药物-基因相互作用数据库中汇编的基因可药性信息。
dgi = drugInteractions(maf = laml, fontSize = 0.75)
dnmt3a.dgi = drugInteractions(genes = "DNMT3A", drugs = TRUE)
## Number of claimed drugs for given genes:
## Gene N
## 1: DNMT3A 7
# Printing selected columns.
dnmt3a.dgi[, .(Gene, interaction_types, drug_name, drug_claim_name)]
## Gene interaction_types drug_name drug_claim_name
## 1: DNMT3A N/A
## 2: DNMT3A DAUNORUBICIN Daunorubicin
## 3: DNMT3A DECITABINE Decitabine
## 4: DNMT3A IDARUBICIN IDARUBICIN
## 5: DNMT3A DECITABINE DECITABINE
## 6: DNMT3A inhibitor DECITABINE CHEMBL1201129
## 7: DNMT3A inhibitor AZACITIDINE CHEMBL1489
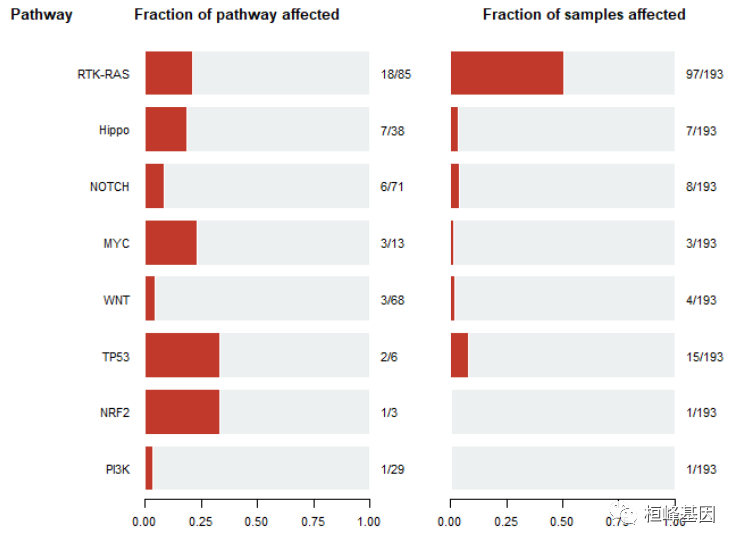
致癌信号通路
在TCGA队列中,致癌途径功能检测已知致癌信号通路的富集,如下:
OncogenicPathways(maf = laml)
## Pathway N n_affected_genes fraction_affected Mutated_samples
## 1: PI3K 29 1 0.03448276 1
## 2: NRF2 3 1 0.33333333 1
## 3: TP53 6 2 0.33333333 15
## 4: WNT 68 3 0.04411765 4
## 5: MYC 13 3 0.23076923 3
## 6: NOTCH 71 6 0.08450704 8
## 7: Hippo 38 7 0.18421053 7
## 8: RTK-RAS 85 18 0.21176471 97
## Fraction_mutated_samples
## 1: 0.005181347
## 2: 0.005181347
## 3: 0.077720207
## 4: 0.020725389
## 5: 0.015544041
## 6: 0.041450777
## 7: 0.036269430
## 8: 0.502590674
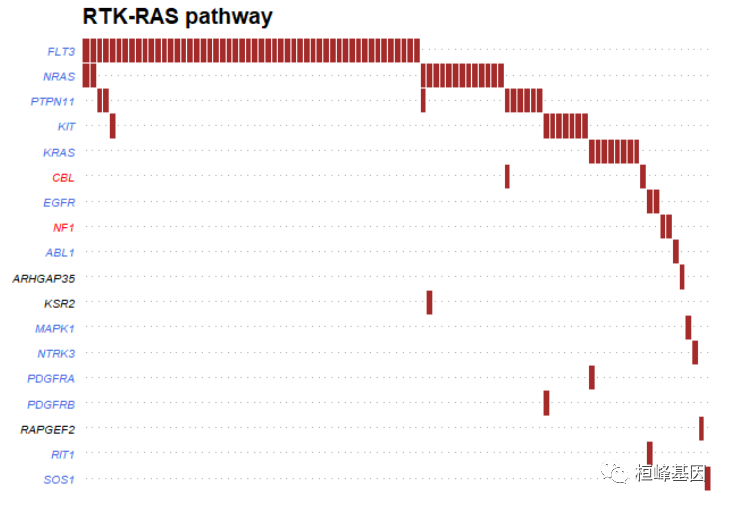
可视化完成的pathway通路,如下:
PlotOncogenicPathways(maf = laml, pathways = "RTK-RAS")
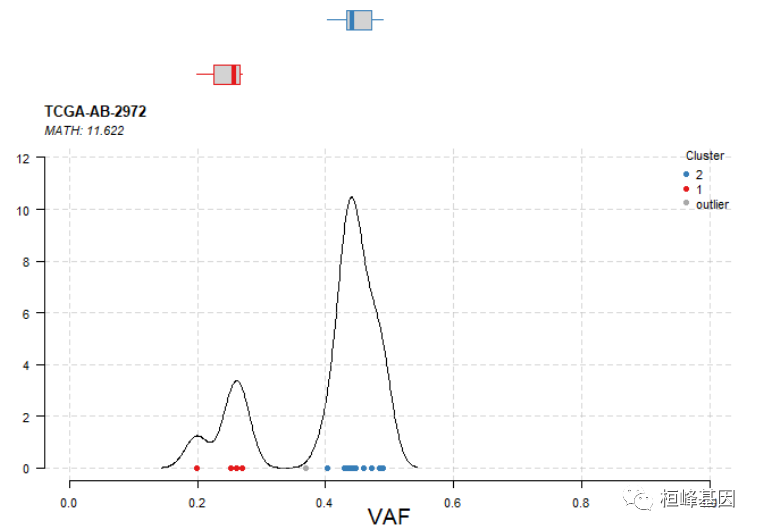
肿瘤样本的异质性
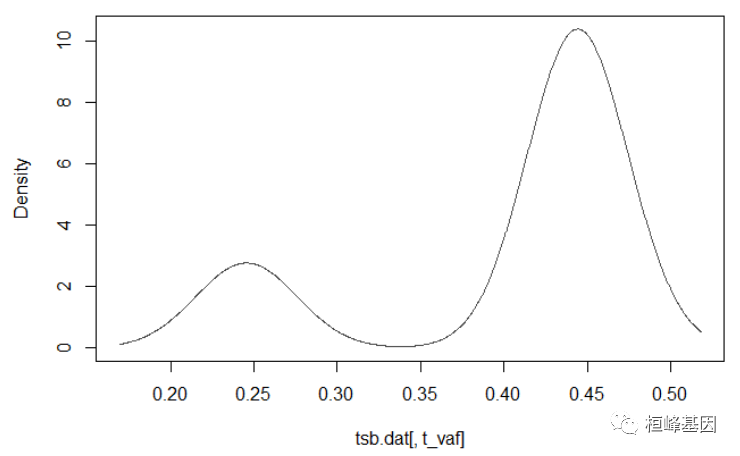
肿瘤通常是异质性的,即由多个克隆组成。这种异质性可以推断集群变异等位基因频率。inferHeterogeneity函数使用vaf信息对变异进行聚类(使用mclust),从而推断克隆性。默认情况下,inferHeterogeneity函数查找包含vaf信息的列t_vaf。但是,如果字段名与t_vaf不同,我们可以使用参数vafCol手动指定。
# Heterogeneity in sample TCGA.AB.2972
library("mclust")
tcga.ab.2972.het = inferHeterogeneity(maf = laml, tsb = "TCGA-AB-2972", vafCol = "i_TumorVAF_WU")
print(tcga.ab.2972.het$clusterMeans)
## Tumor_Sample_Barcode cluster meanVaf
## 1: TCGA-AB-2972 2 0.4496571
## 2: TCGA-AB-2972 1 0.2454750
## 3: TCGA-AB-2972 outlier 0.3695000
可视化结果,如下:
# Visualizing results
plotClusters(clusters = tcga.ab.2972.het)
References:
1.Mayakonda A, Lin DC, Assenov Y, Plass C, Koeffler HP. 2018. Maftools: efficient and comprehensive analysis of somatic variants in cancer. Genome Resarch. PMID: 30341162

















 4091
4091

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









