







点击关注,桓峰基因
桓峰基因公众号推出单细胞系列教程,有需要生信分析的老师可以联系我们!单细胞系列分析教程整理如下:
SCS【4】单细胞转录组数据可视化分析 (Seurat 4.0)
SCS【6】单细胞转录组之细胞类型自动注释 (SingleR)
SCS【7】单细胞转录组之轨迹分析 (Monocle 3) 聚类、分类和计数细胞
SCS【8】单细胞转录组之筛选标记基因 (Monocle 3)
SCS【9】单细胞转录组之构建细胞轨迹 (Monocle 3)
SCS【10】单细胞转录组之差异表达分析 (Monocle 3)
SCS【11】单细胞ATAC-seq 可视化分析 (Cicero)
SCS【12】单细胞转录组之评估不同单细胞亚群的分化潜能 (Cytotrace)
SCS【13】单细胞转录组之识别细胞对“基因集”的响应 (AUCell)
SCS【15】细胞交互:受体-配体及其相互作用的细胞通讯数据库 (CellPhoneDB)
今天来说说细胞交互:受体-配体及其相互作用的细胞通讯数据库 (CellPhoneDB),学会这些分析结果,距离发文章就只差样本的选择了,有创新性的样本将成为文章的亮点,并不是分析内容了!
前 言
介绍
配体受体复合物介导的细胞与细胞之间的通信对于协调各种生物过程,如发育、分化和炎症是至关重要的。为了研究不同细胞类型的环境依赖的串音是如何使生理过程继续进行的,开发了CellPhoneDB,一个新的配体、受体及其相互作用库。与其他知识库相比,我们的数据库考虑了配体和受体的亚基结构,准确地表示了异体复合体。我们将我们的资源与统计框架相结合,从单细胞转录组学数据预测两种细胞类型之间丰富的细胞相互作用。在这里,我们概述了我们的知识库的结构和内容,提供了从单细胞RNA测序数据推断细胞-细胞通信网络的过程,并提供了一个实用的逐步指导来帮助实现该协议。CellPhoneDB v.2.0是包含了额外的功能,使用户能够引入新的相互作用的分子,并减少了查询大型数据集所需的时间和资源。CellPhoneDB v.2.0是公开的,包括代码和用户友好的web界面;为在计算基因组学方面缺乏经验的专家和研究人员使用。例子演示了如何使用已发表的数据集评估与CellPhoneDB v.2.0有意义的生物相互作用。整个流程通常需要2小时来完成,从安装到统计分析和可视化,对于一个约10 GB的数据集,10000个cell和19个cell类型,使用5个线程。
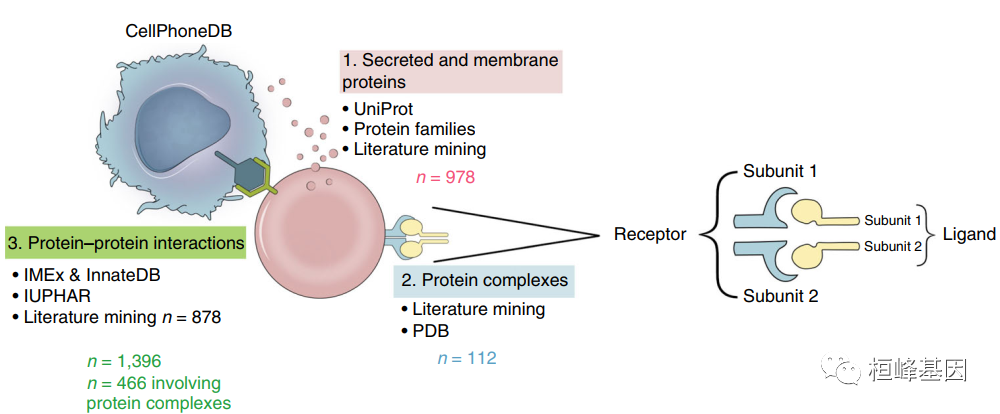
CellPhoneDB是一个公开的受体,配体及其相互作用的资源库。配体和受体都包含亚基结构,准确地表示异体复合物。这是至关重要的,因为细胞与细胞之间的通信依赖于多亚基蛋白质复合体,这超出了大多数数据库和研究中使用的二进制表示。CellPhoneDB集成了与蜂窝通信相关的现有数据集和新的人工审查信息。CellPhoneDB利用了以下数据库的信息:UniProt, ensemble, PDB, IMEx联盟,IUPHAR。CellPhoneDB可用于搜索特定的配体/受体或查询您自己的单细胞转录组学数据。共存储978种蛋白质:其中501种是分泌蛋白,而585种是膜蛋白,这些蛋白质参与1,396种相互作用。CellPhoneDB数据库还考虑了配体和受体的亚基结构,准确地表示了异聚体复合物,有466个是异聚体,对于准确研究多亚基复合物介导的细胞通讯很关键。CellPhoneDB有474种相互作用涉及分泌蛋白,而490种相互作用涉及膜蛋白,总共有250个涉及整合素的相互作用。目前支持的物种:人(也可通过人和鼠的同源基因比对应用于鼠)。
原理
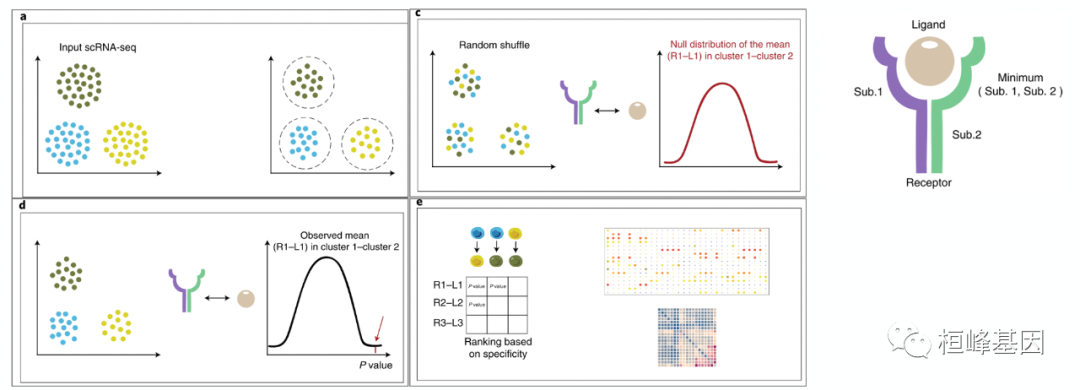
1.CellphoneDB基于一种细胞类型的受体和另一种细胞类型的配体的表达,得出两种细胞群之间丰富的受体-配体相互作用。对于细胞群所表达的基因,计算表达该基因的细胞百分比和基因表达平均值。若该基因只在该群中10%及以下的细胞中表达(默认值为0.1),则被移除。
2.将所有细胞的簇标签随机排列1000次(可选值),并确定簇中受体平均表达水平和相互作用簇中配体平均表达水平的平均值。对于两种细胞类型之间每对比较中的每一个受体-配体对,这产生了一个零分布(null distribution,亦称伯努利分布、两点分布,指的是对于随机变量X有,参数为p(0<p<1),如果它分别以概率p和1-p取1和0为值。EX=p,DX=p(1-p)。伯努利试验成功的次数服从伯努利分布,参数p是试验成功的概率)。
3.通过计算等于或高于实际平均值的平均值的比例,可以得到了一个P值,表示给定受体-配体复合物细胞类型特异性的可能性。换句话说,如果观察到的平均值在前5%,则相互作用的P值为0.05。
4.根据两种细胞类型中富集到的显著的受体-配体对的数量,对细胞类型之间高度特异性的相互作用进行排序,以便手动筛选出生物学相关的相互作用关系。
数据库链接:
https://www.cellphonedb.org/ppi-resources
数据读取
我们需要准备两个文件:
单细胞counts矩阵,行是细胞名称,列是基因,(test_counts.txt)如下:
Gene d-pos_AAACCTGAGCAGGTCA d-pos_AAACCTGGTACCGAGA d-pos_AAACCTGTCGCCATAA d-pos_AAACGGGTCAGTTGAC d-pos_AAAGATGCATTGAGCT d-pos_AAAGATGTCCAAAGTC d-pos_AAAGCAAAGAGGACGG d-pos_AAAGCAACACATTCGA d-pos_AAAGTAGAGAGCCCAA d-pos_AAAGTAGCAAGCTGAG
ENSG00000238009 0 0 0 0 0 0 0 0 0 0
ENSG00000279457 0 0 0 0 0.723769155614119 0 1.1269975757326 1.81828622356148 0 0
ENSG00000228463 0 0 0 0 0 0 0 0 0.737864655764131 1.40825228390187
ENSG00000237094 0 0 0 0 0 0 0 0 0 0
ENSG00000230021 0 0 0 0 0 0 0 0 0 0
ENSG00000237491 0 0 0 0 0 0 0 0 0 0
ENSG00000177757 0 0 0 0 0 0 0 0 0 0
ENSG00000225880 0 0 0 0 0 0 0 0 0 0
ENSG00000230368 0 0 1.06435707230216 0 0 0 0 0 0 0单细胞类型 (test_meta.txt)如下:
1 Cell cell_type
2 d-pos_AAACCTGAGCAGGTCA NKcells_1
3 d-pos_AAACCTGGTACCGAGA NKcells_0
4 d-pos_AAACCTGTCGCCATAA NKcells_1
5 d-pos_AAACGGGTCAGTTGAC Tcells
6 d-pos_AAAGATGCATTGAGCT NKcells_0
7 d-pos_AAAGATGTCCAAAGTC NKcells_0
8 d-pos_AAAGCAAAGAGGACGG Myeloid
9 d-pos_AAAGCAACACATTCGA NKcells_1
10 d-pos_AAAGTAGAGAGCCCAA NKcells_0
11 d-pos_AAAGTAGCAAGCTGAG NKcells_0软件包安装
该软件包是python 的一个包和数据库,因此需要安装python 3.5以上版本,利用conda 安装即可。要使用最新版本,请重新安装cellphonedb并下载数据库:
$pip install -U cellphonedb
$cellphonedb database download当然安装过程也不是一次完成的总是存在很多问题,大部分是python 版本和库的版本问题,报错了自己找一下最新版本的安装即可。
参数说明
整个分析流程就是四个模块进行分析的
database是输入数据库,一般默认可以不输入,直接使用即可;
method:分析模块;
plot :画图模块;
query:进行数据查询的模块;
查询基因有哪些互作结果(get_interaction_gene结果为数据库涉及到的基因,find_interactions_by_element可以找到特定基因的受体配体作用数据对)。
Usage: cellphonedb [OPTIONS] COMMAND [ARGS]...
Options:
--help Show this message and exit.
Commands:
database
method
plot
query
实例操作
$cellphonedb method statistical_analysis test_meta.txt test_counts.txt结果生成四个文件
a. deconvoluted.txt:基因在亚群中的平均表达量
b. means.txt:每对受体-配体的平均表达量
c. pvalues.txt:每对受体-配体的p值
d. significant_means.txt:每对受体-配体显著性结果的平均表达量值
可视化结果:生成三张图形
$cellphonedb plot dot_plot
$cellphonedb plot heatmap_plot cellphonedb_meta.txt
dotplot.pdf CellPhoneDB气泡图:每一列为两个细胞亚群,每一行为一对受体配体名称,颜色代表两个亚群这两个基因的平均表达量高低,越红表示表达越高,气泡大小代表P值的-log10值,气泡越大,说明其越具显著性。
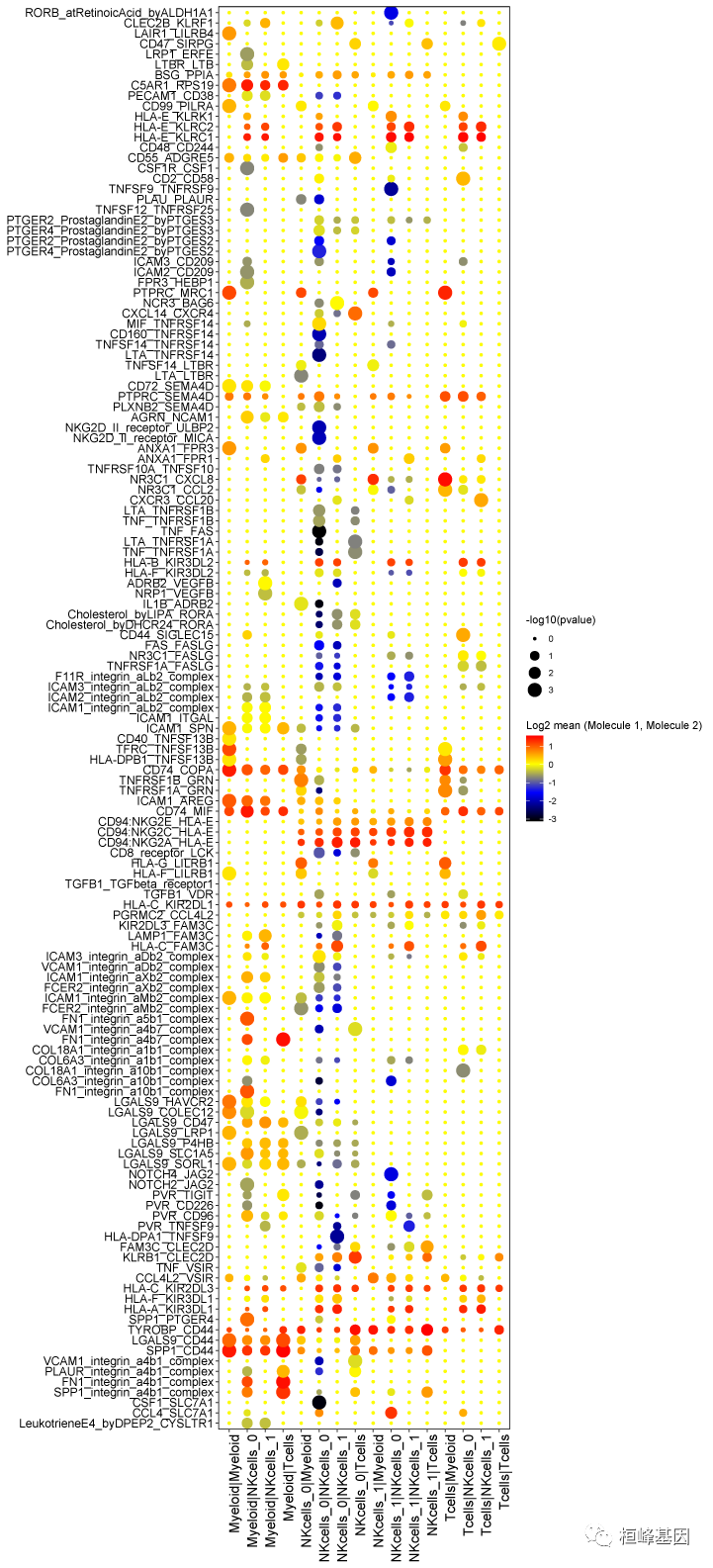
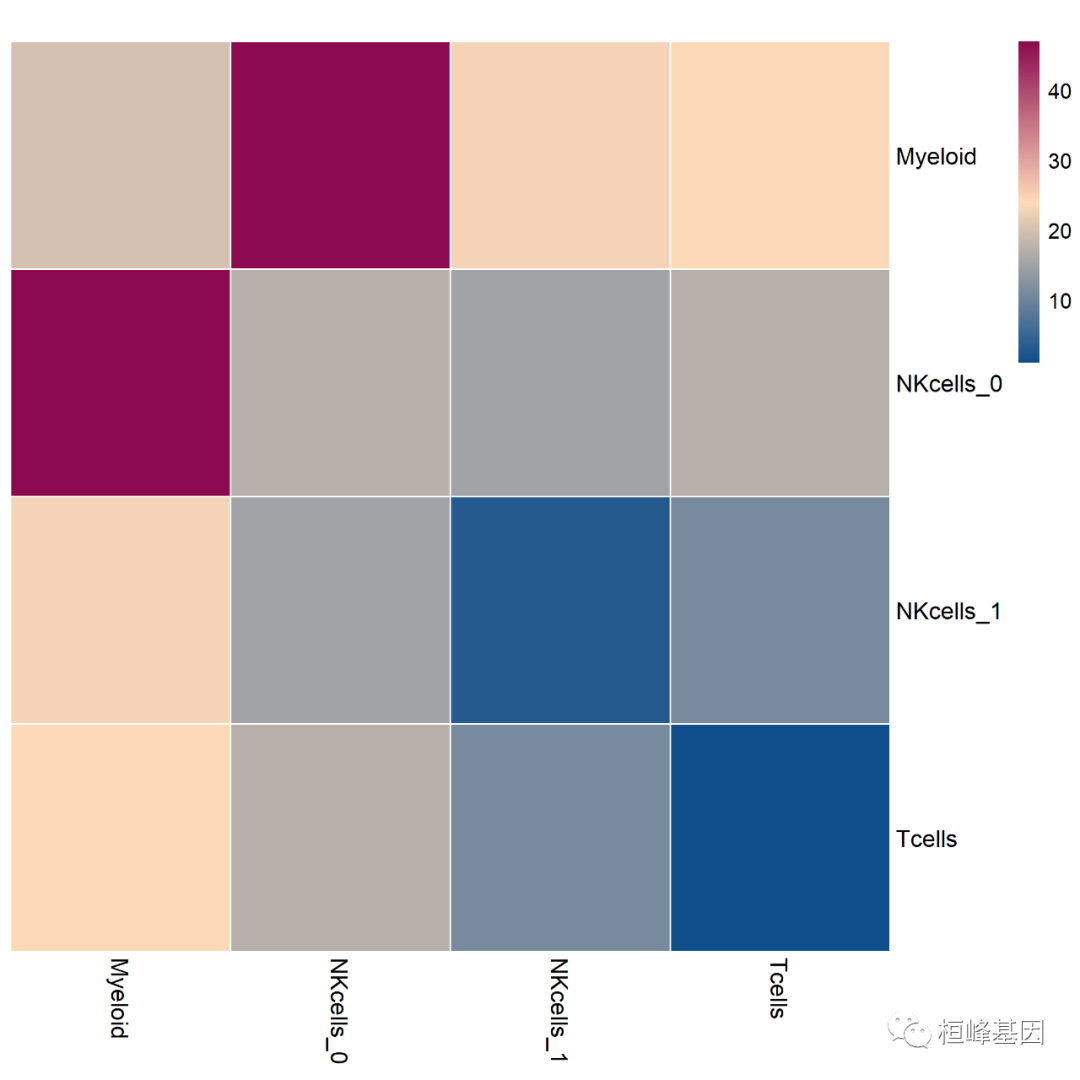
heatmap_count.png和heatmap_log_count.png CellPhoneDB Heatmap:就是对interaction_count.txt结果的展示,下图为亚群之间相互作用受体配体数目的热图:
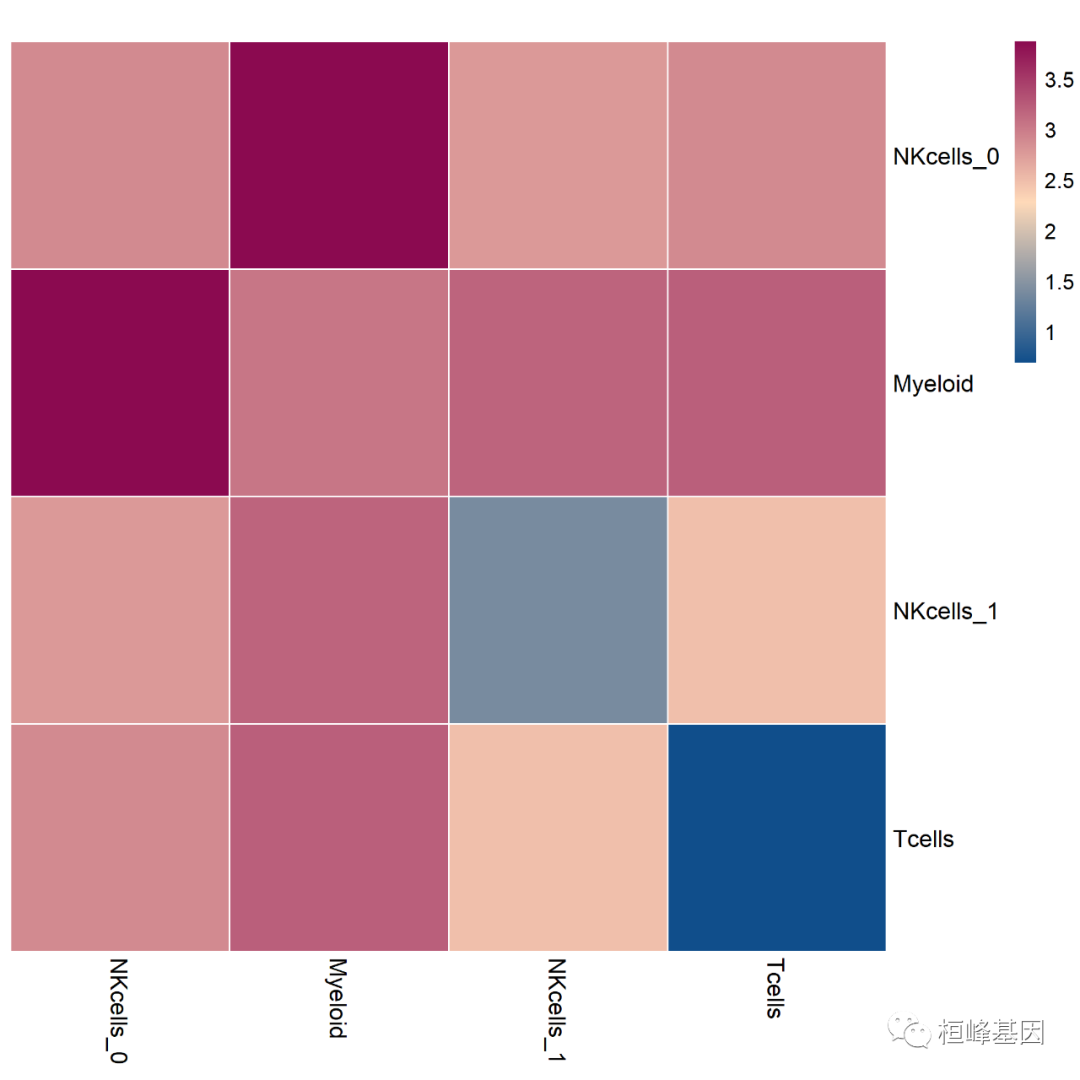
下图为将这个数目取了log10:
若绘图失败,可以到网站上下载R代码自行绘图:
https://github.com/ventolab/CellphoneDB/tree/master/cellphonedb/src/plotters/R
运行命令:
source("plot_dot_by_column_name.R")
dot_plot(means_path = "./out/means.txt", pvalues_path = "./out/pvalues.txt", filename = "./out/dotplot.pdf",
width = 10, height = 22, output_extension = ".pdf")
source("plot_heatmaps.R")
heatmaps_plot(meta_file = "test_meta.txt", pvalues_file = "./out/pvalues.txt", count_filename = "./out/heatmap_counts.png",
log_filename = "./out/heatmap_logcounts.png", count_network_filename = "./out/count_network.txt",
interaction_count_filename = "./out/interactions_count.txt", count_network_separator = "\t",
interaction_count_separator = "\t")结果进行可视化(主要是网络图和点图)
library(psych)
library(qgraph)
library(igraph)
library(tidyverse)
mynet <- read.delim("./out/count_network.txt", check.names = FALSE)
table(mynet$count)
##
## 1 3 11 15 17 20 23 24 47
## 1 1 2 2 3 1 2 2 2
mynet %>%
filter(count > 0) -> mynet # 有零会报错
head(mynet)
## SOURCE TARGET count
## 1 NKcells_1 NKcells_1 3
## 2 NKcells_1 NKcells_0 15
## 3 NKcells_1 Tcells 11
## 4 NKcells_1 Myeloid 23
## 5 NKcells_0 NKcells_1 15
## 6 NKcells_0 NKcells_0 17
net <- graph_from_data_frame(mynet)
plot(net)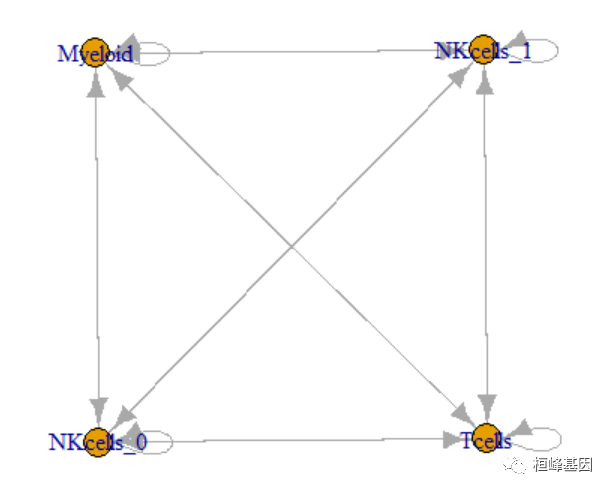
为了给这个网络图的边点mapping上不同的属性我们引入一串颜色
allcolour = c("#DC143C", "#0000FF", "#20B2AA", "#FFA500", "#9370DB", "#98FB98", "#F08080",
"#1E90FF", "#7CFC00", "#FFFF00", "#808000", "#FF00FF", "#FA8072", "#7B68EE",
"#9400D3", "#800080", "#A0522D", "#D2B48C", "#D2691E", "#87CEEB", "#40E0D0",
"#5F9EA0", "#FF1493", "#FFE4B5", "#8A2BE2", "#228B22", "#E9967A", "#4682B4",
"#32CD32", "#F0E68C", "#FFFFE0", "#EE82EE", "#FF6347", "#6A5ACD", "#9932CC",
"#8B008B", "#8B4513", "#DEB887")
karate_groups <- cluster_optimal(net)
coords <- layout_in_circle(net, order = order(membership(karate_groups))) # 设置网络布局
E(net)$width <- E(net)$count/10 # 边点权重(粗细)
plot(net, edge.arrow.size = 0.1, edge.curved = 0, vertex.color = allcolour, vertex.frame.color = "#555555",
vertex.label.color = "black", layout = coords, vertex.label.cex = 0.7)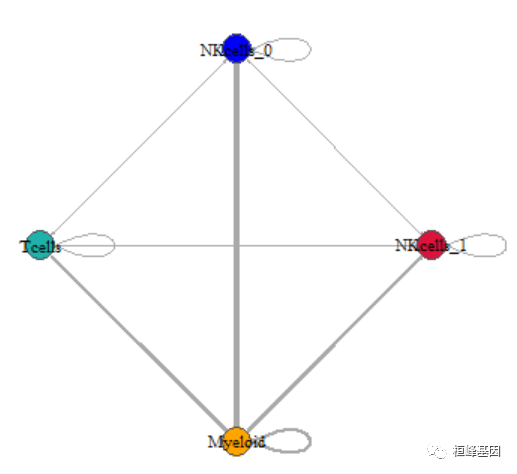
但是边的颜色和点的颜色还是对应不上,我们来修改一番边的属性。
net2 <- net # 复制一份备用
for (i in 1:length(unique(mynet$SOURCE))) {
E(net)[map(unique(mynet$SOURCE), function(x) {
get.edge.ids(net, vp = c(unique(mynet$SOURCE)[i], x))
}) %>%
unlist()]$color <- allcolour[i]
} # 这波操作谁有更好的解决方案?
plot(net, edge.arrow.size = 0.1, edge.curved = 0, vertex.color = allcolour, vertex.frame.color = "#555555",
vertex.label.color = "black", layout = coords, vertex.label.cex = 0.7)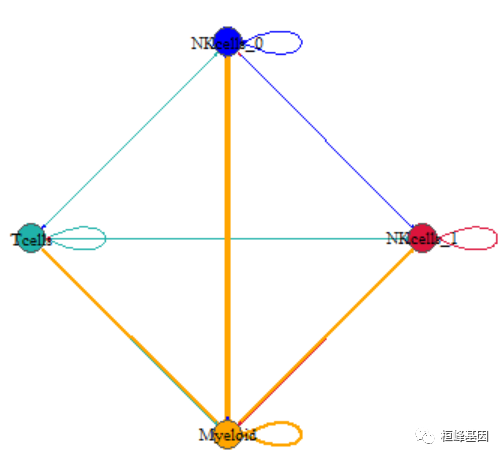
我们观察到由于箭头是双向的,所以两个细胞类型之间的线条会被后来画上去的覆盖掉,这里我们把线条做成曲线:
plot(net, edge.arrow.size=.1,
edge.curved=0.2, # 只是调了这个参数
vertex.color=allcolour,
vertex.frame.color="#555555",
vertex.label.color="black",
layout = coords,
vertex.label.cex=.7)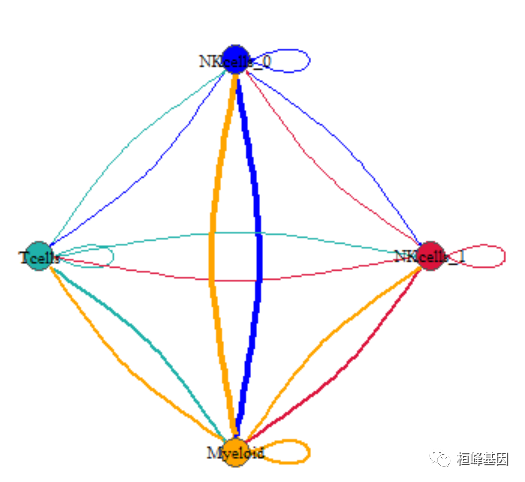
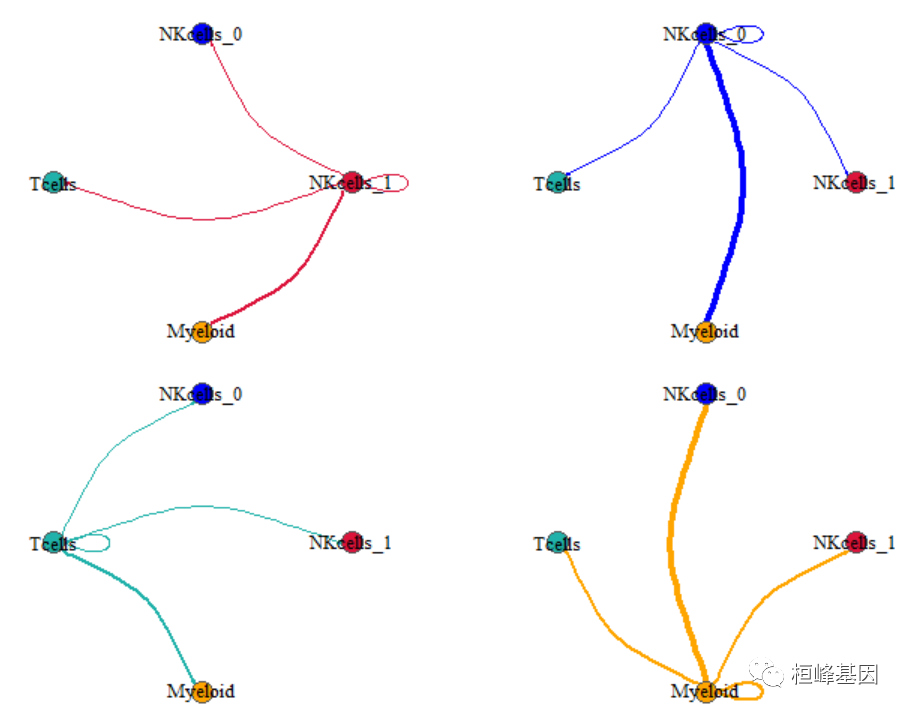
接下来,我们来绘制第二组类型贝壳一样的网络图。
length(unique(mynet$SOURCE)) # 查看需要绘制多少张图,以方便布局
## [1] 4
par(mfrow = c(2, 2), mar = c(0.3, 0.3, 0.3, 0.3))
for (i in 1:length(unique(mynet$SOURCE))) {
net1 <- net2
E(net1)[map(unique(mynet$SOURCE), function(x) {
get.edge.ids(net, vp = c(unique(mynet$SOURCE)[i], x))
}) %>%
unlist()]$color <- allcolour[i]
plot(net1, edge.arrow.size = 0.1, edge.curved = 0.4, vertex.color = allcolour,
vertex.frame.color = "#555555", vertex.label.color = "black", layout = coords,
vertex.label.cex = 1)
}
但是,细胞间通讯的频数(count)并没有绘制在图上,略显不足,这就补上吧。
length(unique(mynet$SOURCE))
## [1] 4
par(mfrow=c(2,2), mar=c(.3,.3,.3,.3))
for (i in 1: length(unique(mynet$SOURCE)) ){
net1<-net2
E(net1)$count <- ""
E(net1)[map(unique(mynet$SOURCE),function(x) {
get.edge.ids(net,vp = c(unique(mynet$SOURCE)[i],x))
})%>% unlist()]$count <- E(net2)[map(unique(mynet$SOURCE),function(x) {
get.edge.ids(net,vp = c(unique(mynet$SOURCE)[i],x))
})%>% unlist()]$count # 故技重施
E(net1)[map(unique(mynet$SOURCE),function(x) {
get.edge.ids(net,vp = c(unique(mynet$SOURCE)[i],x))
})%>% unlist()]$color <- allcolour[i]
plot(net1, edge.arrow.size=.1,
edge.curved=0.4,
edge.label = E(net1)$count, # 绘制边的权重
vertex.color=allcolour,
vertex.frame.color="#555555",
vertex.label.color="black",
layout = coords,
vertex.label.cex=1
)
}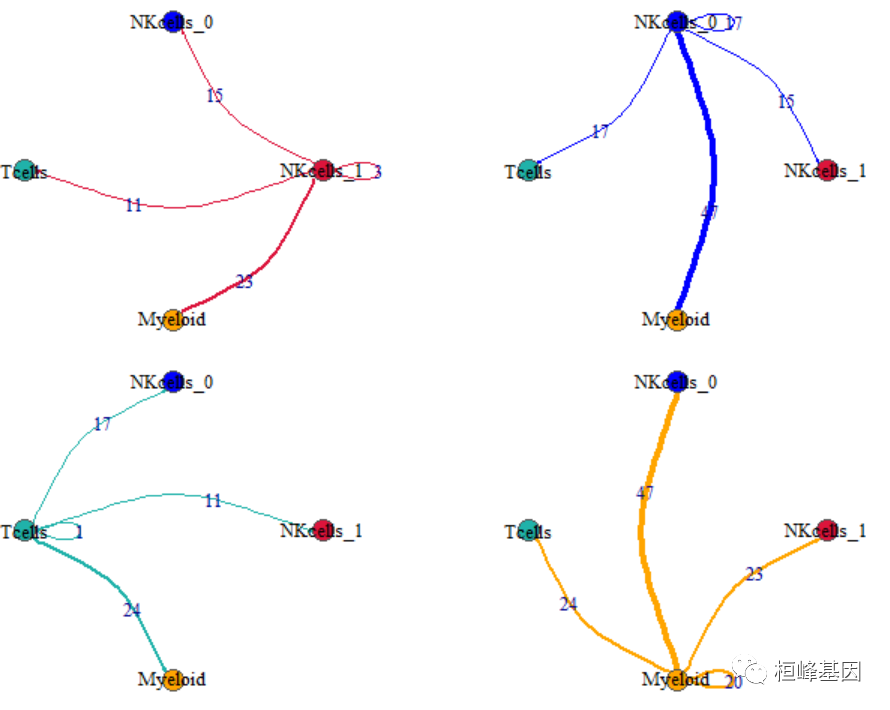
找两条边验证一下对应关系。
mynet %>%
filter(SOURCE == "NKcells_1", TARGET == "Myeloid")
## SOURCE TARGET count
## 1 NKcells_1 Myeloid 23
mynet %>%
filter(SOURCE == "Myeloid", TARGET == "NKcells_0")
## SOURCE TARGET count
## 1 Myeloid NKcells_0 47用ggplot2绘制点图,关键是把两张表合并到一起。
mypvals <- read.delim("./out/pvalues.txt", check.names = FALSE)
mymeans <- read.delim("./out/means.txt", check.names = FALSE)
# 这些基因list很有意思啊,建议保存
chemokines <- grep("^CXC|CCL|CCR|CX3|XCL|XCR", mymeans$interacting_pair, value = T)
chemokines <- grep("^CXC|CCL|CCR|CX3|XCL|XCR", mymeans$interacting_pair, value = T)
th1 <- grep("IL2|IL12|IL18|IL27|IFNG|IL10|TNF$|TNF |LTA|LTB|STAT1|CCR5|CXCR3|IL12RB1|IFNGR1|TBX21|STAT4",
mymeans$interacting_pair, value = T)
th2 <- grep("IL4|IL5|IL25|IL10|IL13|AREG|STAT6|GATA3|IL4R", mymeans$interacting_pair,
value = T)
th17 <- grep("IL21|IL22|IL24|IL26|IL17A|IL17A|IL17F|IL17RA|IL10|RORC|RORA|STAT3|CCR4|CCR6|IL23RA|TGFB",
mymeans$interacting_pair, value = T)
treg <- grep("IL35|IL10|FOXP3|IL2RA|TGFB", mymeans$interacting_pair, value = T)
costimulatory <- grep("CD86|CD80|CD48|LILRB2|LILRB4|TNF|CD2|ICAM|SLAM|LT[AB]|NECTIN2|CD40|CD70|CD27|CD28|CD58|TSLP|PVR|CD44|CD55|CD[1-9]",
mymeans$interacting_pair, value = T)
coinhibitory <- grep("SIRP|CD47|ICOS|TIGIT|CTLA4|PDCD1|CD274|LAG3|HAVCR|VSIR", mymeans$interacting_pair,
value = T)
niche <- grep("CSF", mymeans$interacting_pair, value = T)
mymeans %>%
dplyr::filter(interacting_pair %in% costimulatory) %>%
dplyr::select("interacting_pair", starts_with("NK"), ends_with("NK")) %>%
reshape2::melt() -> meansdf
colnames(meansdf) <- c("interacting_pair", "CC", "means")
mypvals %>%
dplyr::filter(interacting_pair %in% costimulatory) %>%
dplyr::select("interacting_pair", starts_with("NK"), ends_with("NK")) %>%
reshape2::melt() -> pvalsdf
colnames(pvalsdf) <- c("interacting_pair", "CC", "pvals")
pvalsdf$joinlab <- paste0(pvalsdf$interacting_pair, "_", pvalsdf$CC)
meansdf$joinlab <- paste0(meansdf$interacting_pair, "_", meansdf$CC)
pldf <- merge(pvalsdf, meansdf, by = "joinlab")
summary((filter(pldf, means > 1))$means)
## Min. 1st Qu. Median Mean 3rd Qu. Max.
## 1.027 1.292 1.559 1.766 2.300 2.987
pldf %>%
filter(means > 1) %>%
ggplot(aes(CC.x, interacting_pair.x)) + geom_point(aes(color = means, size = -log10(pvals +
1e-04))) + scale_size_continuous(range = c(1, 3)) + scale_color_gradient2(high = "red",
mid = "yellow", low = "darkblue", midpoint = 25) + theme_bw() + theme(axis.text.x = element_text(angle = -45,
hjust = -0.1, vjust = 0.8))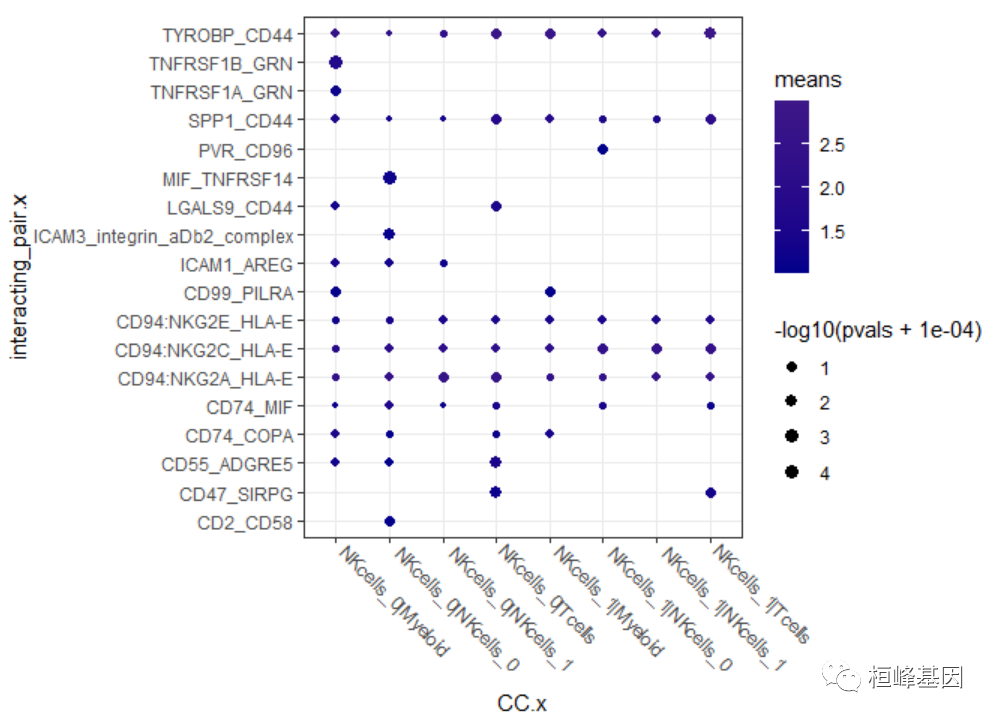
References:
Hie B, Cho H, DeMeo B, Bryson B and Berger B, Geometric Sketching Compactly Summarizes the Single-Cell Transcriptomic Landscape, Cell Systems 2019
CellphonedDBv3: Mapping the temporal and spatial dynamics of the human endometrium in vivo and in vitro. L Garcia-Alonso, L-François Handfield, K Roberts, K Nikolakopoulou et al 2021
CellPhoneDB: Inferring cell-cell communication from combined expression of multi-subunit receptor-ligand complexes Efremova M, Vento-Tormo M, Teichmann S, Vento-Tormo R. Nat Protoc. 2020 Apr;15(4):1484-1506. doi: 10.1038/s41596-020-0292-x. Epub 2020 Feb 26.
Single-cell reconstruction of the early maternal-fetal interface in humans. Vento-Tormo R, Efremova M, et al., Nature. 2018 Nov;563(7731):347-353. doi: 10.1038/s41586-018-0698-6. Epub 2018 Nov 14.
我们这期主要介绍细胞交互:受体-配体及其相互作用的细胞通讯数据库 (CellPhoneDB)。目前单细胞测序的费用也在降低,单细胞系列可算是目前的测序神器,有这方面需求的老师,联系桓峰基因,提供最高端的科研服务!
桓峰基因,铸造成功的您!
未来桓峰基因公众号将不间断的推出单细胞系列生信分析教程,
敬请期待!!
有想进生信交流群的老师可以扫最后一个二维码加微信,备注“单位+姓名+目的”,有些想发广告的就免打扰吧,还得费力气把你踢出去!


















 1039
1039

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









