







简 介
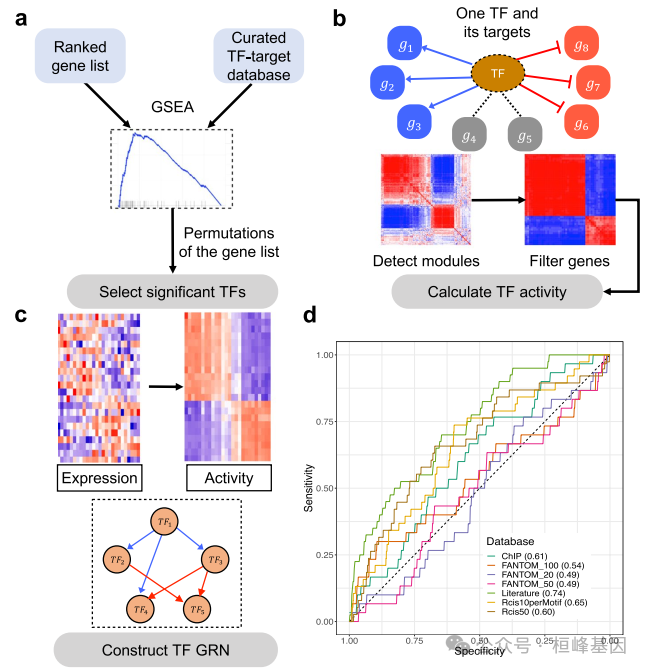
系统生物学的一个主要问题是如何识别控制生物过程决策的核心基因调控回路。一个名为NetAct的计算平台,用于使用转录组学数据和基于文献的转录因子目标数据库构建核心转录因子调控网络。NetAct使用目标表达稳健地推断调控因子的活性,基于转录活性构建网络,并集成数学模型进行验证。我们的计算机基准测试表明,NetAct在推断转录活性和基因网络方面优于 现有算法。我们演示了应用NetAct来模拟驱动TGF-β诱导的上皮-间质转化和巨噬细胞极化的网络。

NetAct是一个计算平台,使用转录组数据和基于文献的转录因子靶点数据库来构建核心转录因子调控网络。本教程旨在演示NetAct的核心功能组件,以及如何使用它来构建和建模转录因子调节网络。本工作流程中使用的实验RNA-seq数据来自Sheridan等人(2015),包括从雌性处女小鼠的乳腺中分选的三个细胞群(基底、腔祖细胞(LP)和成熟腔细胞(ML))。使用GEO:GSE63310,这些样品的count数据可以从Gene Expression Omnibus (GEO)下载。
软件包安装
软件包安装非常简单,但是需要注意一下Rtools,R,BiocManager版本号,这种问题经常出现,更新匹配一直就没问题了。
if(!require(Scissor))
devtools::install_github("lusystemsbio/NetAct")
BiocManager::install("org.Mm.eg.db")数据读取
数据是从GEO上下载,但是这里给了一段代码用于下载数据,但是如果网速很慢还是直接从网站上下载吧:
url <- "https://www.ncbi.nlm.nih.gov/geo/download/?acc=GSE63310&format=file"
utils::download.file(url, destfile="GSE63310_RAW.tar", mode="wb")
utils::untar("GSE63310_RAW.tar", exdir = ".")
files <- c("GSM1545535_10_6_5_11.txt", "GSM1545536_9_6_5_11.txt", "GSM1545538_purep53.txt",
"GSM1545539_JMS8-2.txt", "GSM1545540_JMS8-3.txt", "GSM1545541_JMS8-4.txt",
"GSM1545542_JMS8-5.txt", "GSM1545544_JMS9-P7c.txt", "GSM1545545_JMS9-P8c.txt")
for(i in paste(files, ".gz", sep="")){
R.utils::gunzip(i, overwrite=TRUE)
}对数据进行整理,预处理等操作:
files <- c("GSM1545535_10_6_5_11.txt", "GSM1545536_9_6_5_11.txt", "GSM1545538_purep53.txt",
"GSM1545539_JMS8-2.txt", "GSM1545540_JMS8-3.txt", "GSM1545541_JMS8-4.txt", "GSM1545542_JMS8-5.txt",
"GSM1545544_JMS9-P7c.txt", "GSM1545545_JMS9-P8c.txt")
x <- edgeR::readDGE(files, columns = c(1, 3))
group <- as.factor(c("LP", "ML", "Basal", "Basal", "ML", "LP", "Basal", "ML", "LP"))
x$samples$group <- group
samplenames <- c("LP1", "ML1", "Basal1", "Basal2", "ML2", "LP2", "Basal3", "ML3",
"LP3")
colnames(x) <- samplenames
head(x)
## An object of class "DGEList"
## $samples
## files group lib.size norm.factors
## LP1 GSM1545535_10_6_5_11.txt LP 32863052 1
## ML1 GSM1545536_9_6_5_11.txt ML 35335491 1
## Basal1 GSM1545538_purep53.txt Basal 57160817 1
## Basal2 GSM1545539_JMS8-2.txt Basal 51368625 1
## ML2 GSM1545540_JMS8-3.txt ML 75795034 1
## LP2 GSM1545541_JMS8-4.txt LP 60517657 1
## Basal3 GSM1545542_JMS8-5.txt Basal 55086324 1
## ML3 GSM1545544_JMS9-P7c.txt ML 21311068 1
## LP3 GSM1545545_JMS9-P8c.txt LP 19958838 1
##
## $counts
## Samples
## Tags LP1 ML1 Basal1 Basal2 ML2 LP2 Basal3 ML3 LP3
## 497097 1 2 342 526 3 3 535 2 0
## 100503874 0 0 5 6 0 0 5 0 0
## 100038431 0 0 0 0 0 0 1 0 0
## 19888 0 1 0 0 17 2 0 1 0
## 20671 1 1 76 40 33 14 98 18 8
## 27395 431 771 1368 1268 1564 769 818 468 342实例操作
1. 建立比较和表型数据
library(org.Mm.eg.db)
library(NetAct)
compList <- c("Basal-LP", "Basal-ML", "LP-ML")
phenoData = new("AnnotatedDataFrame", data = data.frame(celltype = group))
rownames(phenoData) = colnames(x$counts)
head(x)
## An object of class "DGEList"
## $samples
## files group lib.size norm.factors
## LP1 GSM1545535_10_6_5_11.txt LP 32863052 1
## ML1 GSM1545536_9_6_5_11.txt ML 35335491 1
## Basal1 GSM1545538_purep53.txt Basal 57160817 1
## Basal2 GSM1545539_JMS8-2.txt Basal 51368625 1
## ML2 GSM1545540_JMS8-3.txt ML 75795034 1
## LP2 GSM1545541_JMS8-4.txt LP 60517657 1
## Basal3 GSM1545542_JMS8-5.txt Basal 55086324 1
## ML3 GSM1545544_JMS9-P7c.txt ML 21311068 1
## LP3 GSM1545545_JMS9-P8c.txt LP 19958838 1
##
## $counts
## Samples
## Tags LP1 ML1 Basal1 Basal2 ML2 LP2 Basal3 ML3 LP3
## 497097 1 2 342 526 3 3 535 2 0
## 100503874 0 0 5 6 0 0 5 0 0
## 100038431 0 0 0 0 0 0 1 0 0
## 19888 0 1 0 0 17 2 0 1 0
## 20671 1 1 76 40 33 14 98 18 8
## 27395 431 771 1368 1268 1564 769 818 468 3422. 预处理和DEG分析
对于RNA-seq数据,NetAct提供了一个有用的预处理函数,称为preprocess_counts(),可以过滤掉低表达的基因,并为原始计数数据检索相关的基因。然后使用limma/DESeq2包鉴定差异表达基因(DEG)。生成的规范化表达式数据与phenoData元数据一起保存到标准ExpressionSet对象中,以便进行下游分析。
limma
counts <- Preprocess_counts(counts = x$counts, groups = group, mouse = TRUE)
DErslt = RNAseqDegs_limma(counts = counts, phenodata = phenoData, complist = compList,
qval = 0.05)
## Basal-LP Basal-ML LP-ML
## Down 4308 4568 2570
## NotSig 7071 6728 11506
## Up 4882 4965 2185
neweset = Biobase::ExpressionSet(assayData = as.matrix(DErslt$Overall$e), phenoData = phenoData)DESeq2
DErslt2 = RNAseqDegs_DESeq(counts = counts, phenodata = phenoData, complist = compList,
qval = 0.05)
neweset2 = Biobase::ExpressionSet(assayData = as.matrix(DErslt2$Overall$e), phenoData = phenoData)3. 鉴定显著富集的TF
采用置换的方法,通过GSEA算法选择显著富集的TF。通过考虑q值截止值,从多个比较中汇总TF。此步骤通常耗时最长,因此建议通过提供文件名来保存输出。
data("mDB")
calc <- FALSE
if (calc) {
gsearslts <- TF_Selection(GSDB = mDB, DErslt = DErslt, minSize = 5, nperm = 10000,
qval = 0.05, compList = compList, nameFile = "gsearslts_tutorial")
} else {
gsearslts <- readRDS(file = "gsearslts_tutorial.RDS")
}
tfs <- gsearslts$tfs
tfs
## [1] "Ar" "Cebpa" "Chd7" "Ctnnb1" "E2f1" "Egr1" "Esr1" "Ets1"
## [9] "Foxo1" "Foxo3" "Gli1" "Gli2" "Hnf1a" "Hoxc8" "Lef1" "Myocd"
## [17] "Myod1" "Nfatc1" "Nfkb1" "Npas2" "Pgr" "Pou2f1" "Ppara" "Pparg"
## [25] "Sfn" "Smad3" "Smad4" "Sox2" "Sp1" "Sp3" "Srf" "Tfap2a"
## [33] "Trp53"
Reselect_TFs(GSEArslt = gsearslts$GSEArslt, qval = 0.01)
## [1] "Ar" "Cebpa" "Ctnnb1" "Foxo1" "Foxo3" "Gli1" "Gli2" "Myocd"
## [9] "Myod1" "Pou2f1" "Ppara" "Pparg" "Smad4" "Sp1" "Tfap2a"4. 计算所选TF的活性
NetAct根据其目标的表达来推断所选监管机构的活动。结果显示与继承基因表达相比,TF活性模式更清晰。
act.me <- TF_Activity(tfs, mDB, neweset, DErslt$Overall)
acts_mat = act.me$all_activities
Activity_heatmap(acts_mat, neweset)
5. 构建基因调控网络
在这一步中,通过基于互信息和熵的链路关系过滤,输出基因调控网络拓扑结构。plot_network()函数提供网络拓扑的交互式视图。
tf_links = TF_Filter(acts_mat, mDB, miTh = 0.05, nbins = 8, corMethod = "spearman",
DPI = T)
## [1] 0
## [1] 0.5
plot_network(tf_links)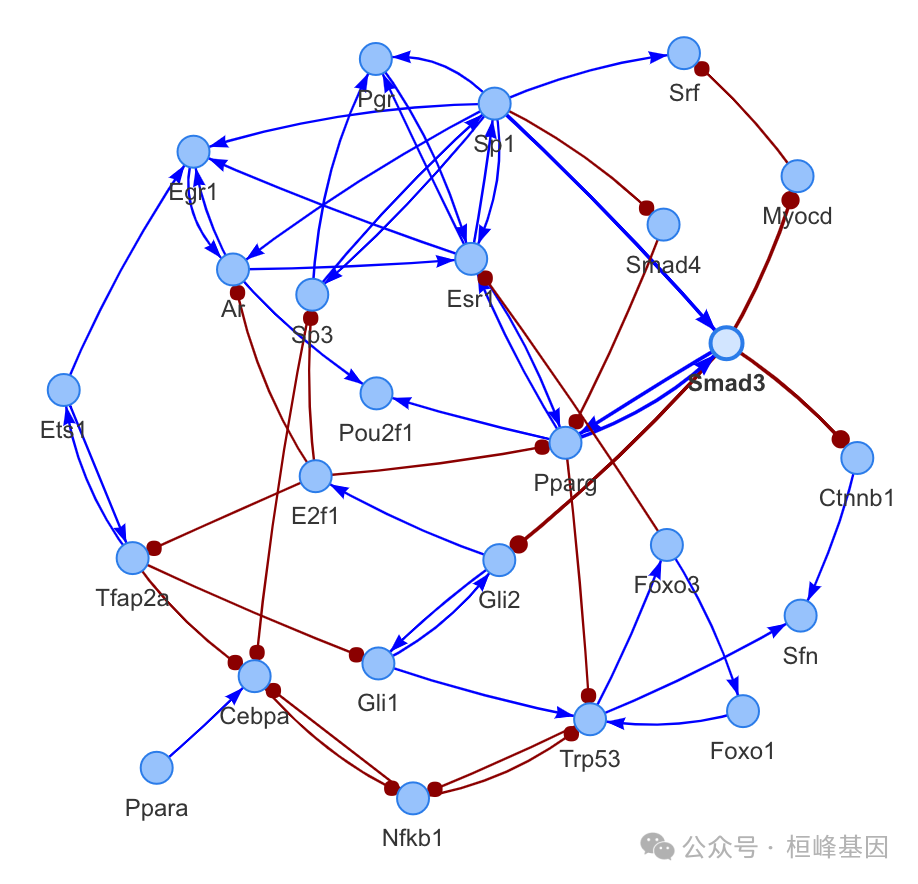
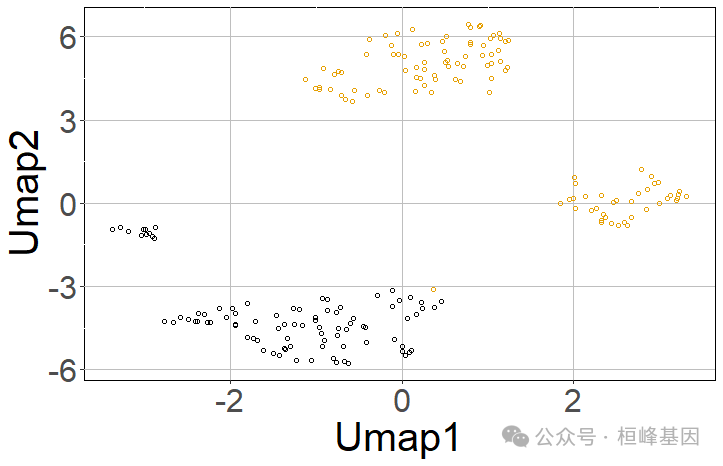
6. RACIPE模拟
然后,使用RACIPE算法对构建的网络的动态行为进行建模。建议模拟10000个模型。
racipe_results <- sRACIPE::sracipeSimulate(circuit = tf_links, numModels = 200, plots = TRUE)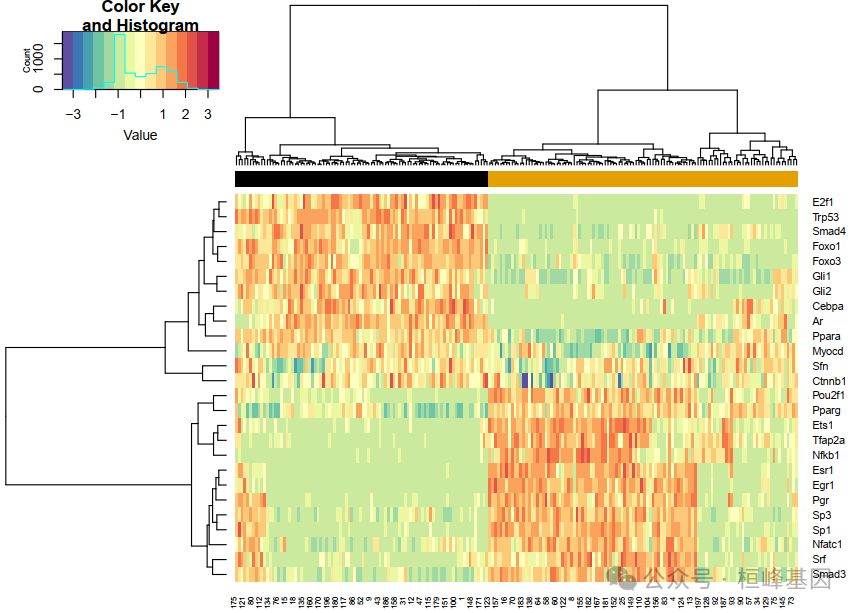
Reference
Su K, et al (2022) NetAct: a computational platform to construct core transcription factor regulatory networks using gene activity, Genome Biology, 23:270.
桓峰基因,铸造成功的您!
未来桓峰基因公众号将不间断的推出单细胞系列生信分析教程,
敬请期待!!
桓峰基因官网正式上线,请大家多多关注,还有很多不足之处,大家多多指正!http://www.kyohogene.com/
桓峰基因和投必得合作,文章润色优惠85折,需要文章润色的老师可以直接到网站输入领取桓峰基因专属优惠券码:KYOHOGENE,然后上传,付款时选择桓峰基因优惠券即可享受85折优惠哦!https://www.topeditsci.com/


















 9812
9812

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









