








接上文:STAR比hisat2更好;hisat2快一些得花几个小时
featureCount是subread软件包里的一个命令,所以安装subread即可。
官网(Subread download | SourceForge.net)下载subread-2.0.3-Linux-x86_64.tar.gz压缩文件
wget https://sourceforge.net/projects/subread/files/subread-2.0.6/subread-2.0.6-Linux-x86_64.tar.gz/download
tar -zxvf进行解压,进入bin/目录,目录下featureCounts就可直接运行
#找到featureCount位置:/home/data/t210451/miniconda3/pkgs/subread-2.0.6-Linux-x86_64/bin

#每次/临时添加环境变量:
export PATH=/home/data/t210451/miniconda3/pkgs/subread-2.0.6-Linux-x86_64/bin:$PATH
#下载注释文件gencode.v29.annotation.gtf.gz
#查看当前文件夹下所有bam文件
ls -lh /home/data/t210451/RNAseq/fhdata/fhclean/trim-galore/fastp/aligned/bam/*.bam
dirlist=$(ls -t /home/data/t210451/RNAseq/fhdata/fhclean/trim-galore/fastp/aligned/bam/*.bam |tr '\n' ' ')
echo $dirlist
featureCounts -a /home/data/t210451/RNAseq/fhdata/fhclean/aligned/bam/gencode.v29.annotation.gtf.gz -o /home/data/t210451/RNAseq/fhdata/fhclean/trim-galore/fastp/aligned/bam/6_final_counts.txt -g 'gene_name' -T 8 -p $dirlist
报错:Paired-end reads were detected in single-end read library——加-p
https://www.reddit.com/r/bioinformatics/comments/13m89bd/help_with_featurecounts_error_pairedend_reads/?rdt=47295#从featureCounts输出文件中获取counts与TPM矩阵 RNA-seq入门实战(三):在R里面整理表达量counts矩阵-腾讯云开发者社区-腾讯云 (tencent.com)
####有个问题
cd /home/data/t210451/RNAseq/zzdata/zzclean/fastp/aligned/sambam
#进入生成的6_final_counts.txt.summary文件夹所在目录
multiqc 6_final_counts.txt.summary
#整合后看匹配的reads比例和数量
file:///D:/%E8%B0%B7%E6%AD%8C%E4%B8%8B%E8%BD%BD/multiqc_report.html
#不对劲,特别少
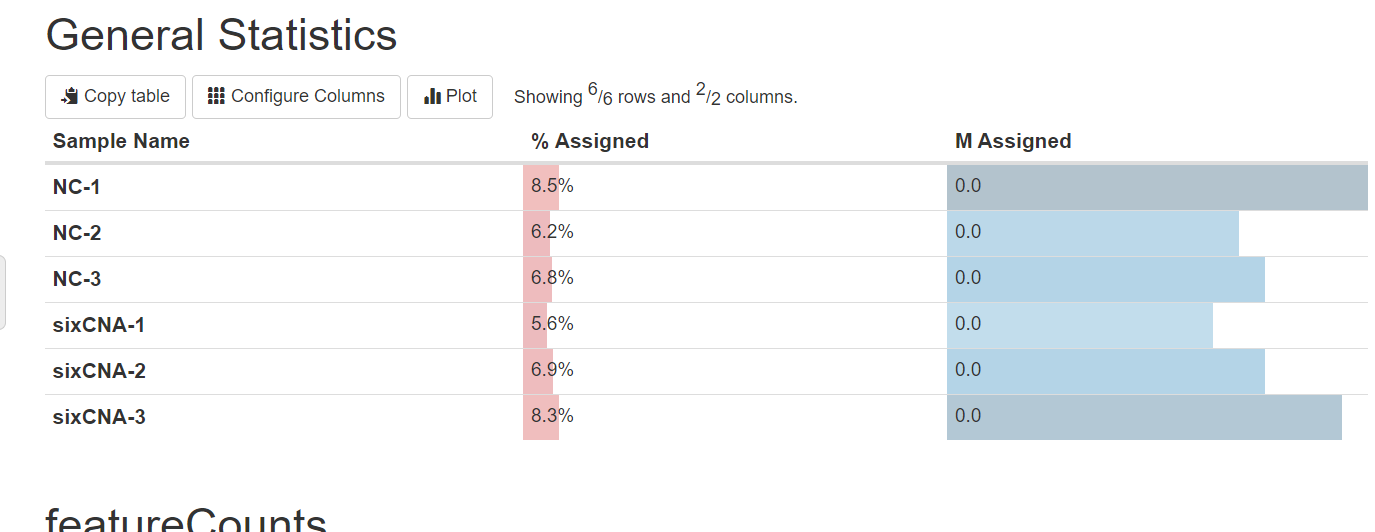
#发现featureCounts比对的GRCh38是human的!!!!gencode.v29.annotation.gtf.gz

### 重新下载注释文件并且重新比对(老鼠的应该是GRCm38=mm10)
#下载mouse的注释文件:GENCODE - Mouse Release M33

#上传后重新比对,看multiqc结果

#增加了一些,但不多。。。。。哪里出问题了呢?之前,我的参考基因文件(我是在ncbi下载的)和注释文件(genecode网站)下载的? 保姆级参考基因组及其注释下载方法(图文详解) - 知乎 (zhihu.com)
在同一个网站下载fasta

1、 读取counts.txt构建counts矩阵;
2、样品的重命名和分组;
3、counts与TPM转换;
4、基因ID转换;
5、初步过滤低表达基因与保存counts数据
















 7568
7568











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









