







三种最常用的差异分析方法(deseq2,edgeR,limma_voom)的整理。
目前在实际应用的过程中一般选择其中一种结果即可,或三种方法分析后结果取交集。

本次演示选择了GSE213615数据集,该数据集采用了两种肝癌细胞系,并使用索拉菲尼处理,最后得到了索拉菲尼耐药细胞,差异分析的目的是观察索拉菲尼耐药组相比于对照组而言的肝癌细胞基因变化情况。
差异分析前数据准备
1、导入数据并处理
rm(list = ls())
library(dplyr)
proj = "GSE213615"
# Raw-data已经被研究者所清洗,合并即可
file_directory = "~/desktop/dat/GSE213615_RAW" #设置路径
fs=list.files(file_directory) #列出路径下所有文件
exp = do.call(cbind,lapply(fs, function(x){
# 读取文件
a <- read.table(file.path(file_directory, x),
header = T, sep = '\t', row.names = NULL,
quote = "",comment.char = "")
# 去除lncRNA数据
a <- a[!grepl("lncRNA", a$description),]
# 提取含有 "Hep" 或 "Huh" 字样的列和 "symbol" 列
selected_cols <- which(grepl("Hep|Huh", colnames(a)))
selected_cols <- c(selected_cols, which(colnames(a) == "symbol"))
a <- a[, selected_cols]
# 提取文件名并去除扩展名
file_name <- strsplit(basename(x), "_")[[1]][1]
# 对列名进行重命名,使用文件名作为前缀
colnames(a)[colnames(a) != "symbol"] <- file_name
# 返回处理后的数据框
return(a)
}))
exp[1:4,1:4]
# 这里do.call函数的作用是对后面的lapply函数中得到的数据进行cbind处理。
# lapply函数的作用是将fs中的每一个文件进行自定义函数处理,这里就是读取每一个文件。
exp <- distinct(exp, symbol, .keep_all = T)
rownames(exp) <- exp$symbol
exp <- exp[, !grepl("symbol", colnames(exp))]
2、提取临床信息
library(GEOquery)
eSet = getGEO("GSE213615",getGPL = F,destdir = ".")
eSet = eSet[[1]]
clinical = pData(eSet)
#提取自己需要的
colnames(clinical)
clinical = clinical[,c(1:2,47:48)]
3、基因过滤
#请注意这里必须是count数据!!!
#需要过滤一下那些在很多样本里表达量都为0或者表达量很低的基因。过滤标准不唯一。
#过滤之前基因数量:
nrow(exp)
# 仅保留在一半以上样本里表达的基因
exp = exp[apply(exp, 1, function(x) sum(x > 0) > 0.5*ncol(exp)), ]
nrow(exp)
exp =round(exp)
4、获取分组信息
k = stringr::str_detect(clinical$`treatment:ch1`,"None");table(k)
Group = ifelse(k,"control","resistent")
Group = factor(Group,levels = c("control","resistent"))
table(Group)
5、重新对clinical和exp数据进行排序整理
p = identical(rownames(clinical),colnames(exp));p
if(!p) {
s = intersect(rownames(clinical),colnames(exp))
exp = exp[,s]
clinical = clinical[s,]
}
6、保存数据
#差异基因分析是count值,实在不行可以用tpm
save(exp,Group,proj,clinical,file = paste0(proj,".Rdata"))
deseq2差异分析
rm(list = ls())
options(stringsAsFactors = F)
library(ggplot2)
library(ggstatsplot)
library(ggsci)
library(RColorBrewer)
library(patchwork)
library(ggplotify)
library(limma)
library(edgeR)
library(DESeq2)
proj = "GSE213615"
load(file = paste0(proj,".Rdata"))
# 这里的赋值是为了方便后面的分析过程
exprSet <- exp
group_list <- Group
# 制作分组表格
colData <- data.frame(row.names=colnames(exprSet),
group_list=group_list)
# 基因表达值变成整数
exprSet <- apply(exprSet, 2, as.integer)
rownames(exprSet) <- rownames(exp)
# 构建分析数据,exprSet,colData和group_list的排序必须一致
dds <- DESeqDataSetFromMatrix(countData = exprSet,
colData = colData,
design = ~ group_list)
dds <- DESeq(dds)
# 这里的group_list需因子化
res <- results(dds,
contrast=c("group_list",
levels(group_list)[2],
levels(group_list)[1]))
# contrast函数规定了比较的顺序,是前者比后者,在这里就是resistent组比control组
levels(group_list)[2];levels(group_list)[1]
# [1] "resistent"
# [1] "control"
# 按照padj的顺序进行从小到大排序
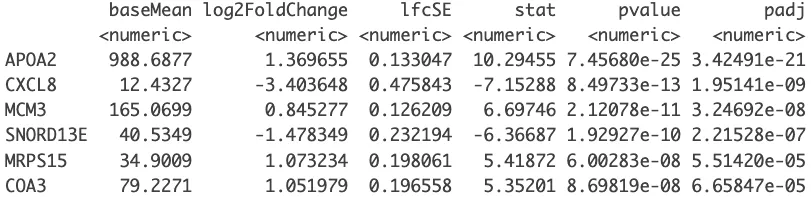
resOrdered <- res[order(res$padj),]
head(resOrdered)
DEG =as.data.frame(resOrdered)
DEG_deseq2 = na.omit(DEG)
save(DEG_deseq2,
file = 'DEG_deseq2.Rdata' )
edgeR差异分析
proj = "GSE213615"
load(file = paste0(proj,".Rdata"))
# 这里的赋值是为了方便后面的分析过程
exprSet <- exp
group_list <- Group
# 使用 DGEList 函数创建一个边缘回归(edgeR)的数据对象
d <- DGEList(counts=exprSet,
group= group_list)
# 使用cpm计算每个基因的Counts Per Million (CPM) 值。然后筛选出在至少两个样本中 CPM 大于1的基因,以过滤掉低表达的基因
keep <- rowSums(cpm(d)>1) >= 2
table(keep)
d <- d[keep, , keep.lib.sizes=FALSE]
# 重新计算每个样本的库大小(库中的总读数),并更新 d$samples 中的库大小信息。
d$samples$lib.size <- colSums(d$counts)
# 计算并应用标准化因子(Normalization Factors),这有助于在不同样本之间进行公平的比较。
d <- calcNormFactors(d)
d$samples
dge=d
# 创建无截距(intercept)的模型矩阵,将每个分组视为单独的模型参数
design <- model.matrix(~0+factor(group_list))
rownames(design)<-colnames(dge)
colnames(design)<-levels(factor(group_list))
# 估计所有基因的共有离散度(common dispersion)
dge <- estimateGLMCommonDisp(dge,design)
# 估计基因表达水平随平均表达变化的趋势性离散度(trended dispersion)
dge <- estimateGLMTrendedDisp(dge, design)
# 估计每个基因特异性的离散度(tagwise dispersion),即对每个基因单独进行离散度估计
dge <- estimateGLMTagwiseDisp(dge, design)
# 根据设计矩阵 design 使用广义线性模型(GLM)拟合数据对象 dge,并返回拟合结果 fit
fit <- glmFit(dge, design)
# https://www.biostars.org/p/110861/
#使用广义线性模型对比两个组(这里的对比是第二组相对于第一组,即 contrast=c(-1, 1)),计算出每个基因的似然比检验(Likelihood Ratio Test)统计量
lrt <- glmLRT(fit, contrast=c(-1,1))
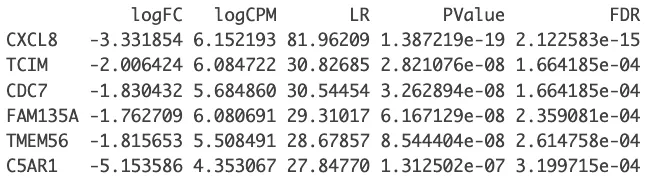
# 从 lrt 结果中提取前 n 个基因(这里 n = nrow(dge) 表示提取所有基因)的差异表达结果
nrDEG=topTags(lrt, n=nrow(dge))
DEG_edgeR = as.data.frame(nrDEG)
head(DEG_edgeR)
save(DEG_edgeR,
file = 'DEG_edgeR.Rdata' )
limma-voom
proj = "GSE213615"
load(file = paste0(proj,".Rdata"))
# 这里的赋值是为了方便后面的分析过程
exprSet <- exp
group_list <- Group
# 创建无截距的设计矩阵,其中每个分组作为单独的模型参数
design <- model.matrix(~0+factor(group_list))
colnames(design)=levels(factor(group_list))
rownames(design)=colnames(exprSet)
design
# control resistent
# GSM6589876 0 1
# GSM6589877 0 1
# GSM6589878 0 1
# GSM6589879 1 0
# GSM6589880 1 0
# GSM6589881 1 0
# GSM6589882 0 1
# GSM6589883 0 1
# GSM6589884 0 1
# GSM6589885 1 0
# GSM6589886 1 0
# GSM6589887 1 0
# attr(,"assign")
# [1] 1 1
# attr(,"contrasts")
# attr(,"contrasts")$`factor(group_list)`
# [1] "contr.treatment"
# 使用DGEList函数创建一个edgeR的数据对象dge
dge <- DGEList(counts=exprSet)
# 计算并应用标准化因子(Normalization Factors)
dge <- calcNormFactors(dge)
# 计算Counts Per Million(CPM)值,并取对数变换
logCPM <- cpm(dge, log=TRUE, prior.count=3)
# voom 函数将RNA-seq数据转换为类似微阵列数据的格式,以便应用线性模型进行分析
v <- voom(dge,design,plot=TRUE, normalize="quantile")
# 使用线性模型拟合经过 voom 处理的数据 "v"
fit <- lmFit(v, design)
group_list
g1=levels(group_list)[1]
g2=levels(group_list)[2]
# 创建对比组字符串,表示将比较 g2 与 g1 的差异表达
con=paste0(g2,'-',g1)
cat(con)
# 创建一个对比矩阵,用于指定要比较的组别。con 是之前创建的对比字符串
cont.matrix=makeContrasts(contrasts=c(con),levels = design)
# 应用对比矩阵到线性模型 fit 中,得到 fit2
fit2=contrasts.fit(fit,cont.matrix)
# 使用经验贝叶斯方法对模型 fit2 进行调整,以提高统计精度
fit2=eBayes(fit2)
# 提取差异表达分析的结果
# n=Inf 表示提取所有基因。coef=con表示基于对比con提取结果。
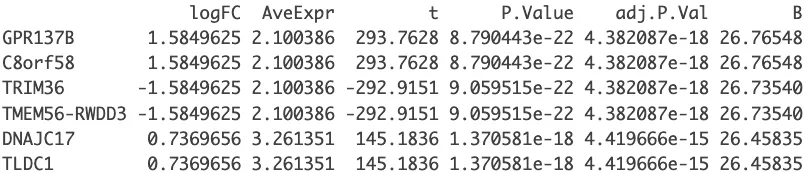
tempOutput = topTable(fit2, coef=con, n=Inf)
DEG_limma_voom = na.omit(tempOutput) # 去除结果中的缺失值(NA)。
head(DEG_limma_voom)
save(DEG_limma_voom,
file = 'DEG_limma_voom.Rdata' )
参考资料:
1、deseq2:https://bioconductor.org/packages/devel/bioc/vignettes/DESeq2/inst/doc/DESeq2.html
2、edgeR:https://bioconductor.org/packages/release/bioc/vignettes/edgeR/inst/doc/edgeRUsersGuide.pdf
3、limma-voom: https://ucdavis-bioinformatics-training.github.io/2018-June-RNA-Seq-Workshop/thursday/DE.html
4、生信技能树:https://mp.weixin.qq.com/s/G7LQHvybW32Kn-jPYR7k6A
致谢:感谢曾老师,小洁老师以及生信技能树团队全体成员。
注:若对内容有疑惑或者有发现明确错误的朋友,请联系后台(欢迎交流)。更多内容可关注公众号:生信方舟
- END -

















 4102
4102

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









