










 该文通过第一性原理分子动力学和Marcus理论研究了Al掺杂MgO与水界面的电子行为。发现多余电子主要形成局域电子和氢自由基,分析了电子转移至这两种状态的活化势垒,揭示了光解水低效率的原因——水合电子而非氢自由基为主导的反应途径。
该文通过第一性原理分子动力学和Marcus理论研究了Al掺杂MgO与水界面的电子行为。发现多余电子主要形成局域电子和氢自由基,分析了电子转移至这两种状态的活化势垒,揭示了光解水低效率的原因——水合电子而非氢自由基为主导的反应途径。
题目:Pathways for Electron Transfer at MgO–Water Interfaces from Ab Initio Molecular Dynamics
作者:Zhutian Ding、Zachary K. Goldsmith和Annabella Selloni*
文章大意:金属氧化物–水界面上的电子转移是催化方面的重要问题,了解电子转移的详细机制和电子转移与氢气生成之间的关系具有极为重要的意义。
本文利用基于杂化密度泛函理论的分子动力学并结合Marcus电子转移理论(有时间写写关于Marcus电子转移理论的内容),研究了金属氧化物–水界面处的电子行为。模拟结果显示,氧化镁中掺铝引入的多余电子将会被局域在一个带隙中间的缺陷态上,这个缺陷态的能量和形状与电子水合物中的电子态差不多。我们观察到两种副产品均与这个多余电子有关:一个是界面上的局域电子,另一个是液体中的氢自由基。
从能量的角度看,电子更趋向于形成氢自由基,此时反应的能量会降到最低。但转变为界面局域电子的的势垒更低。计算得到的两个反应方向活化势垒的能量差与实验观察到的结果一致,即水合电子才是水受光激发生产氢气的主要物质,而不是氢自由基。这些结果为MgO光解水的低效率提供了解释。
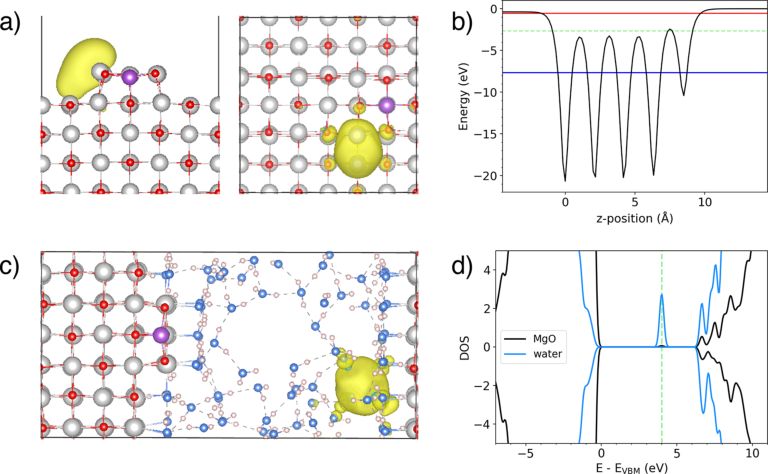
图1. (a)Al掺杂MgO(001)界面及其真空层;(b)垂直于界面方向的势能;(c)Al掺杂MgO-水界面示意图;(d)界面上MgO和水的态密度。
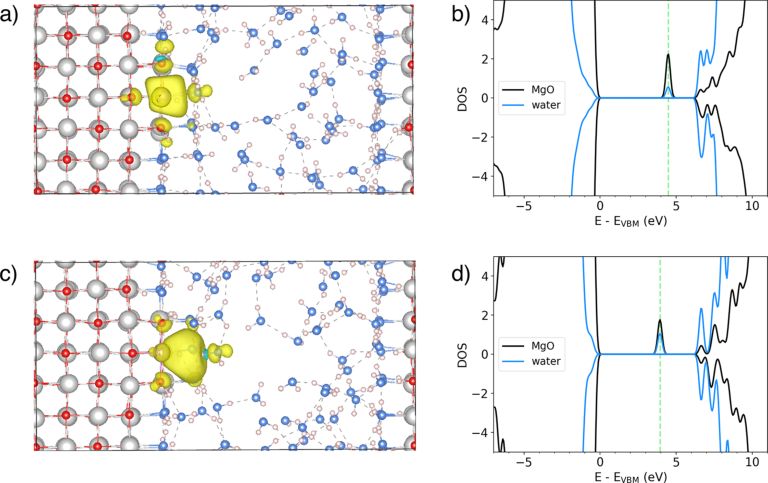
图2. 初始时刻(a)和终止时刻(c)界面上多余电子活动的路径,(b)和(d)为相应的态密度
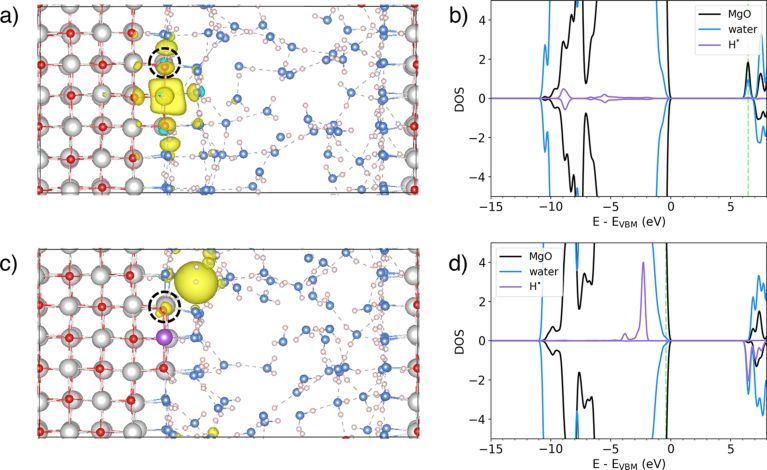
图3. 初始时刻(a)和终止时刻(c)界面上氢自由基活动的路径,(b)和(d)为相应的态密
度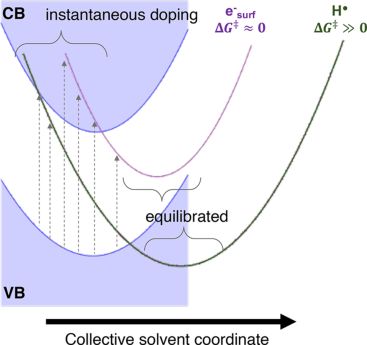
图4. 电子从MgO转移为界面上局域电子或氢自由基的路径。


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









