







 文章介绍了如何使用snpEff软件对gatk产生的SNP.vcf文件进行注释,包括构建物种数据库、执行注释命令以及解析结果文件。snpEff提供了多种参数选项,如上下游长度、输出格式和过滤条件等,用于定制化分析。注释后的vcf文件可用于进一步的SNP下游分析,如4DTv位点分析和进化树构建。
文章介绍了如何使用snpEff软件对gatk产生的SNP.vcf文件进行注释,包括构建物种数据库、执行注释命令以及解析结果文件。snpEff提供了多种参数选项,如上下游长度、输出格式和过滤条件等,用于定制化分析。注释后的vcf文件可用于进一步的SNP下游分析,如4DTv位点分析和进化树构建。
目录
利用snpEff软件对 snp.vcf (利用gatk软件calling-snp)进行注释,运行下述命令:
## 构建好物种的数据库
java -jar /opt/snpEff/snpEff.jar build -c ./snpEff.config -gtf22 -v Ath10M
## 进行注释
java -jar /opt/snpEff/snpEff.jar -c ./snpEff.config -ud 5000 -csvStats test.csv -htmlStats test.html -o vcf Ath10M ../data/all.filtered.snp.vcf > all.filtered.snp.ann.vcf
1.帮助文档
java -jar /data/home/hgzhong/home/software_set/snpeff/snpEff/snpEff.jar --help
##参数
Options:
-chr <string> : Prepend 'string' to chromosome name (e.g. 'chr1' instead of '1'). Only on TXT output.
-classic : Use old style annotations instead of Sequence Ontology and Hgvs.
-csvStats <file> : Create CSV summary file.
-download : Download reference genome if not available. Default: true
-i <format> : Input format [ vcf, bed ]. Default: VCF.
-fileList : Input actually contains a list of files to process.
-o <format> : Ouput format [ vcf, gatk, bed, bedAnn ]. Default: VCF.
-s , -stats, -htmlStats : Create HTML summary file. Default is 'snpEff_summary.html'
-noStats : Do not create stats (summary) file
Results filter options:
-fi , -filterInterval <file> : Only analyze changes that intersect with the intervals specified in this file (you may use this option many times)
-no-downstream : Do not show DOWNSTREAM changes
-no-intergenic : Do not show INTERGENIC changes
-no-intron : Do not show INTRON changes
-no-upstream : Do not show UPSTREAM changes
-no-utr : Do not show 5_PRIME_UTR or 3_PRIME_UTR changes
-no <effectType> : Do not show 'EffectType'. This option can be used several times.
Annotations options:
-cancer : Perform 'cancer' comparisons (Somatic vs Germline). Default: false
-cancerSamples <file> : Two column TXT file defining 'oringinal \t derived' samples.
-fastaProt <file> : Create an output file containing the resulting protein sequences.
-formatEff : Use 'EFF' field compatible with older versions (instead of 'ANN').
-geneId : Use gene ID instead of gene name (VCF output). Default: false
-hgvs : Use HGVS annotations for amino acid sub-field. Default: true
-hgvsOld : Use old HGVS notation. Default: false
-hgvs1LetterAa : Use one letter Amino acid codes in HGVS notation. Default: false
-hgvsTrId : Use transcript ID in HGVS notation. Default: false
-lof : Add loss of function (LOF) and Nonsense mediated decay (NMD) tags.
-noHgvs : Do not add HGVS annotations.
-noLof : Do not add LOF and NMD annotations.
-noShiftHgvs : Do not shift variants according to HGVS notation (most 3prime end).
-oicr : Add OICR tag in VCF file. Default: false
-sequenceOntology : Use Sequence Ontology terms. Default: true
Generic options:
-c , -config : Specify config file
-configOption name=value : Override a config file option
-d , -debug : Debug mode (very verbose).
-dataDir <path> : Override data_dir parameter from config file.
-download : Download a SnpEff database, if not available locally. Default: true
-nodownload : Do not download a SnpEff database, if not available locally.
-h , -help : Show this help and exit
-noLog : Do not report usage statistics to server
-q , -quiet : Quiet mode (do not show any messages or errors)
-v , -verbose : Verbose mode
-version : Show version number and exit
Database options:
-canon : Only use canonical transcripts.
-canonList <file> : Only use canonical transcripts, replace some transcripts using the 'gene_id transcript_id' entries in <file>.
-interaction : Annotate using inteactions (requires interaciton database). Default: true
-interval <file> : Use a custom intervals in TXT/BED/BigBed/VCF/GFF file (you may use this option many times)
-maxTSL <TSL_number> : Only use transcripts having Transcript Support Level lower than <TSL_number>.
-motif : Annotate using motifs (requires Motif database). Default: true
-nextProt : Annotate using NextProt (requires NextProt database).
-noGenome : Do not load any genomic database (e.g. annotate using custom files).
-noExpandIUB : Disable IUB code expansion in input variants
-noInteraction : Disable inteaction annotations
-noMotif : Disable motif annotations.
-noNextProt : Disable NextProt annotations.
-onlyReg : Only use regulation tracks.
-onlyProtein : Only use protein coding transcripts. Default: false
-onlyTr <file.txt> : Only use the transcripts in this file. Format: One transcript ID per line.
-reg <name> : Regulation track to use (this option can be used add several times).
-ss , -spliceSiteSize <int> : Set size for splice sites (donor and acceptor) in bases. Default: 2
-spliceRegionExonSize <int> : Set size for splice site region within exons. Default: 3 bases
-spliceRegionIntronMin <int> : Set minimum number of bases for splice site region within intron. Default: 3 bases
-spliceRegionIntronMax <int> : Set maximum number of bases for splice site region within intron. Default: 8 bases
-strict : Only use 'validated' transcripts (i.e. sequence has been checked). Default: false
-ud , -upDownStreamLen <int> : Set upstream downstream interval length (in bases)
1.1 常用参数
-ud, updownStreamLen :距离SNP位点上下游的长度
java -jar: Java环境下运行程序
-c,-config: snpEff.config配置文件路径(此处的是将其复制至当前目录)
-gff3 :设置输入基因组注释信息是gff3格式,如果是gtf文件请改为-gtf22
-v ,-verbose:设置在程序运行过程中输出的日志信息,设置输入的基因组版本信息,和snpEff.config配置文件中添加的信息一致
-o :输出文件格式
2. 命令的用法:
Usage: snpEff [eff] [options] genome_version [input_file]
##例子
java -jar /opt/snpEff/snpEff.jar -c ./snpEff.config -ud 5000 -csvStats test.csv -htmlStats test.html -o vcf Ath10M ../data/all.filtered.snp.vcf > all.filtered.snp.ann.vcf3. 结果文件解读
获得的结果文件:
test.csv
test.html
test.genes.txt
all.filtered.snp.ann.vcf*.ann.vcf 是一个注释结果文件,其就在vcf的info信息新添加了anno一列信息,其具体每个值含义如下:
Allele
突变之后的碱基,第一个突变位点由T碱基突变成了C碱基,对应Allel的值为C
Annotation
由sequence ontology定义的突变类型
Annotation_Impact
对变异位点有害程度的简单评估,取值有HIGH, MODERATE, LOW, MODIFIER 4种,含义如下

查看 test.genes.txt 文件:
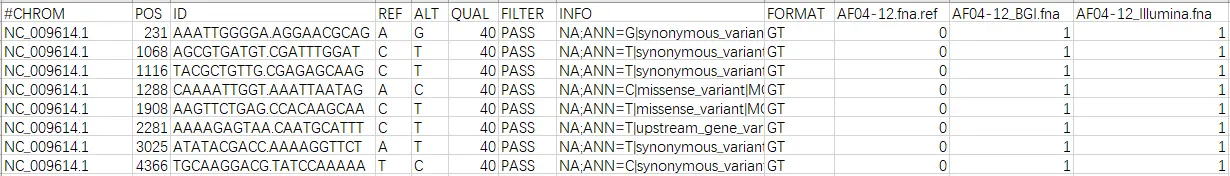
1 染色体
2 突变位置
3 突变周边,“.”的右边时突变位置
4 参考碱基
5 突变碱基
6 是否通过过滤
7 突变类型,氨基酸变化,上下游、基因间、内含子的突变情况
8 参考时0
9/10 不突变0,突变1
第七列详情,任取三个突变
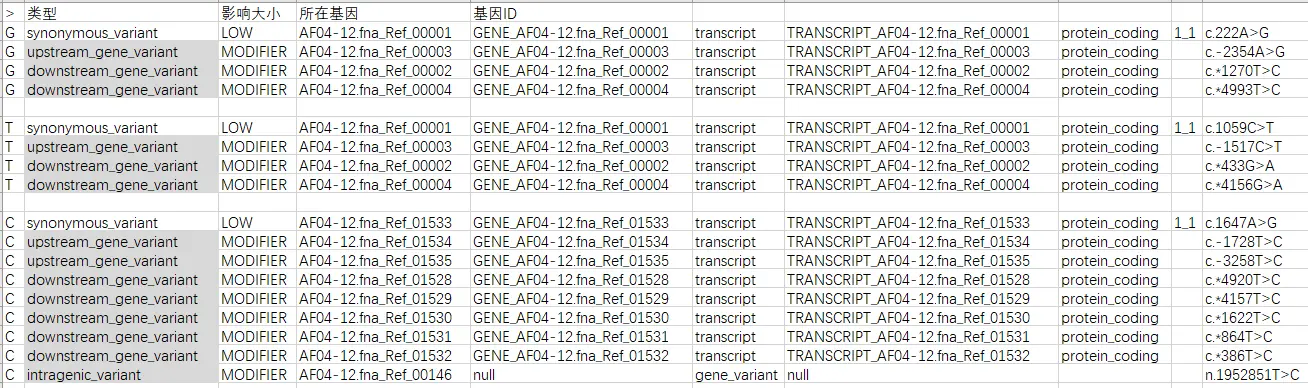
结果 test.html 文件:
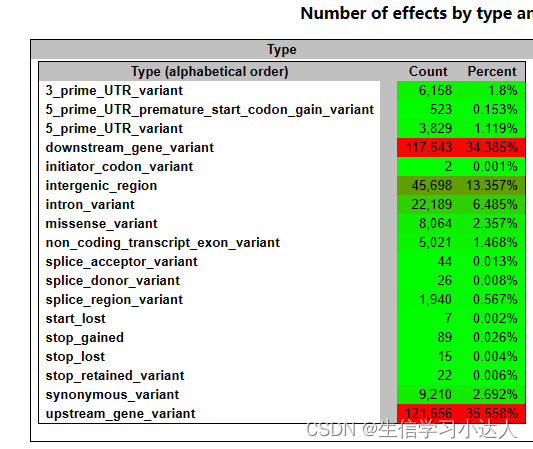

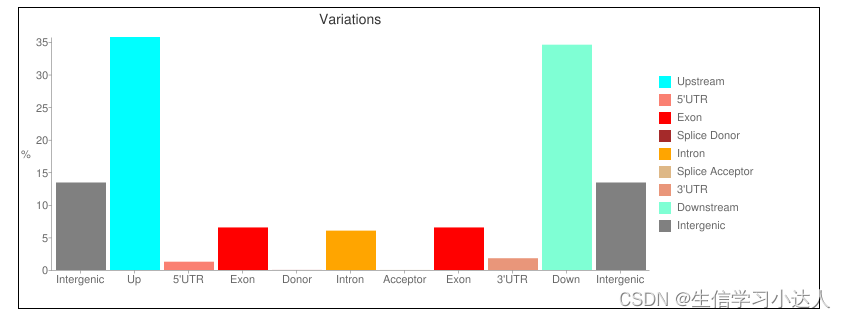
注释结果的HTML文件的详情解读请参照 snpeff结果解读
4. SNP下游的分析
可以使用snpEff注释的vcf进行4DTv位点分析,然后用其构建进化树。
或者是直接使用vcf构建进化树。
两种方法构建进化树均已经实现流程自动化。Vcf2Tree github

















 8881
8881

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









