









一、涉及的新概念
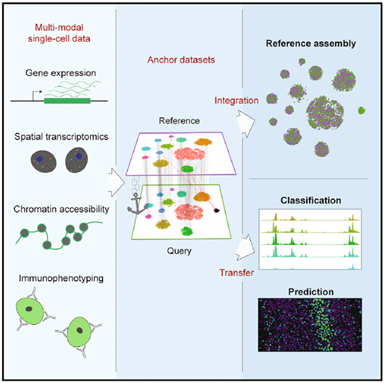
参考(reference):将跨个体,跨技术,跨模式产生的不同的单细胞数据整合后的数据集 。也就是将不同来源的数据集组合到同一空间(reference)中。 从广义上讲,在概念上类似于基因组DNA序列的参考装配。
查询(query):单个实验产生的数据集
转化学习(transfer learning):产生一个于参考数据集(reference)上进行训练的模型,可以将信息再重新投影到query datase上
锚定:由一组共同的分子特征定义的两个细胞(每个数据集一个),将对应关系表示锚定。将得到的一对细胞为锚点,它们编码的跨数据集的细胞关系,将构成所有后续整合分析的基础。
二、标准流程
安装数据集
library(Seurat)
library(SeuratData)
InstallData("panc8")
这里如果长时间下载不了,尝试以下的方法:
-
可以在Rstudio的控制台看到下载链接,将它复制到本地下载:https://seurat.nygenome.org/src/contrib/panc8.SeuratData_3.0.2.tar.gz

-
待下载完成,解压,将标注文件复制出来

-
复制到R环境的库目录,比如我的是:E:\R\R-3.6.1\library\SeuratData\data
数据预处理
rm(list = ls())
options(stringsAsFactors = F)
library(Seurat)
library(SeuratData)
data("panc8")
pancreas.list <- SplitObject(panc8, split.by = "tech")
pancreas.list <- pancreas.list[c("celseq", "celseq2", "fluidigmc1", "smartseq2")]
# 先对数据集进行归一化,并为每个识别位点确定可变特征。
# 特征选择方法使用variance stabilizing transformation ("vst")
for (i in 1:length(pancreas.list)) {
pancreas.list[[i]] <- NormalizeData(pancreas.list[[i]], verbose = FALSE)
pancreas.list[[i]] <- FindVariableFeatures(pancreas.list[[i]], selection.method = "vst",
nfeatures = 2000, verbose = FALSE)
}
整合数据集
# 整合3种测序方法的胰岛细胞数据集
reference.list <- pancreas.list[c("celseq", "celseq2", "smartseq2")]
# 识别锚点
# 这里选的维度是30,作者建议可以在10-50间调试
pancreas.anchors <- FindIntegrationAnchors(object.list = reference.list, dims = 1:30)
# 进行数据集整合
# 已经整合后的表达矩阵存储在Assay中,未处理的表达举证在RNA对象中
pancreas.integrated <- IntegrateData(anchorset = pancreas.anchors, dims = 1:30)
可视化
library(ggplot2)
library(cowplot)
DefaultAssay(pancreas.integrated) <- "integrated"
pancreas.integrated <- ScaleData(pancreas.integrated, verbose = FALSE)
pancreas.integrated <- RunPCA(pancreas.integrated, npcs = 30, verbose = FALSE)
pancreas.integrated <- RunUMAP(pancreas.integrated, reduction = "pca", dims = 1:30)
p1 <- DimPlot(pancreas.integrated, reduction = "umap", group.by = "tech")
p2 <- DimPlot(pancreas.integrated, reduction = "umap", group.by = "celltype", label = TRUE,
repel = TRUE) + NoLegend()
plot_grid(p1, p2)
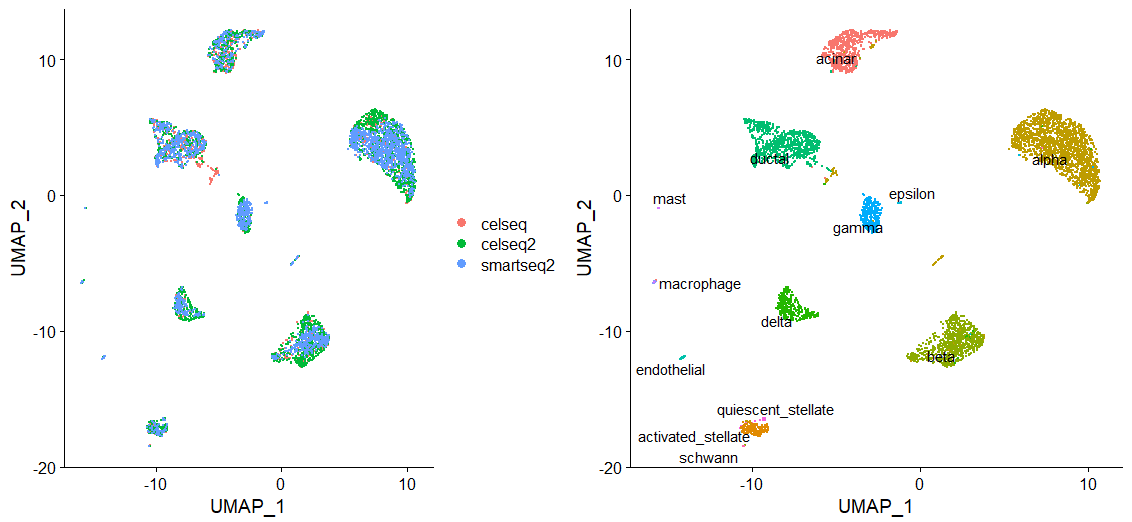
使用装配参考数据集进行细胞类型分类
三、SCTransform 流程
rm(list = ls())
options(stringsAsFactors = F)
library(Seurat)
library(ggplot2)
options(future.globals.maxSize = 4000 * 1024^2)
data("panc8")
数据预处理
pancreas.list <- SplitObject(panc8, split.by = "tech")
pancreas.list <- pancreas.list[c("celseq", "celseq2", "fluidigmc1", "smartseq2")]
# 对每个项目运行SCTransform
for (i in 1:length(pancreas.list)) {
pancreas.list[[i]] <- SCTransform(pancreas.list[[i]], verbose = FALSE)
}
# 接下来,为下游分析选择特征,运行 PrepSCTIntegration, 确保已计算出所有必要的Pearson
pancreas.features <- SelectIntegrationFeatures(object.list = pancreas.list, nfeatures = 3000)
pancreas.list <- PrepSCTIntegration(object.list = pancreas.list, anchor.features = pancreas.features, verbose = FALSE)
整合数据集
# 这里选择归一化方法为“SCT”,其他命令与标准化流程一样
pancreas.anchors <- FindIntegrationAnchors(object.list = pancreas.list, normalization.method = "SCT",
anchor.features = pancreas.features, verbose = FALSE)
pancreas.integrated <- IntegrateData(anchorset = pancreas.anchors, normalization.method = "SCT",
verbose = FALSE)
细胞分群
pancreas.integrated <- RunPCA(pancreas.integrated, verbose = FALSE)
pancreas.integrated <- RunUMAP(pancreas.integrated, dims = 1:30)
plots <- DimPlot(pancreas.integrated, group.by = c("tech", "celltype"), combine = FALSE)
plots <- lapply(X = plots, FUN = function(x) x + theme(legend.position = "top") + guides(color = guide_legend(nrow = 3, byrow = TRUE, override.aes = list(size = 3))))
CombinePlots(plots)
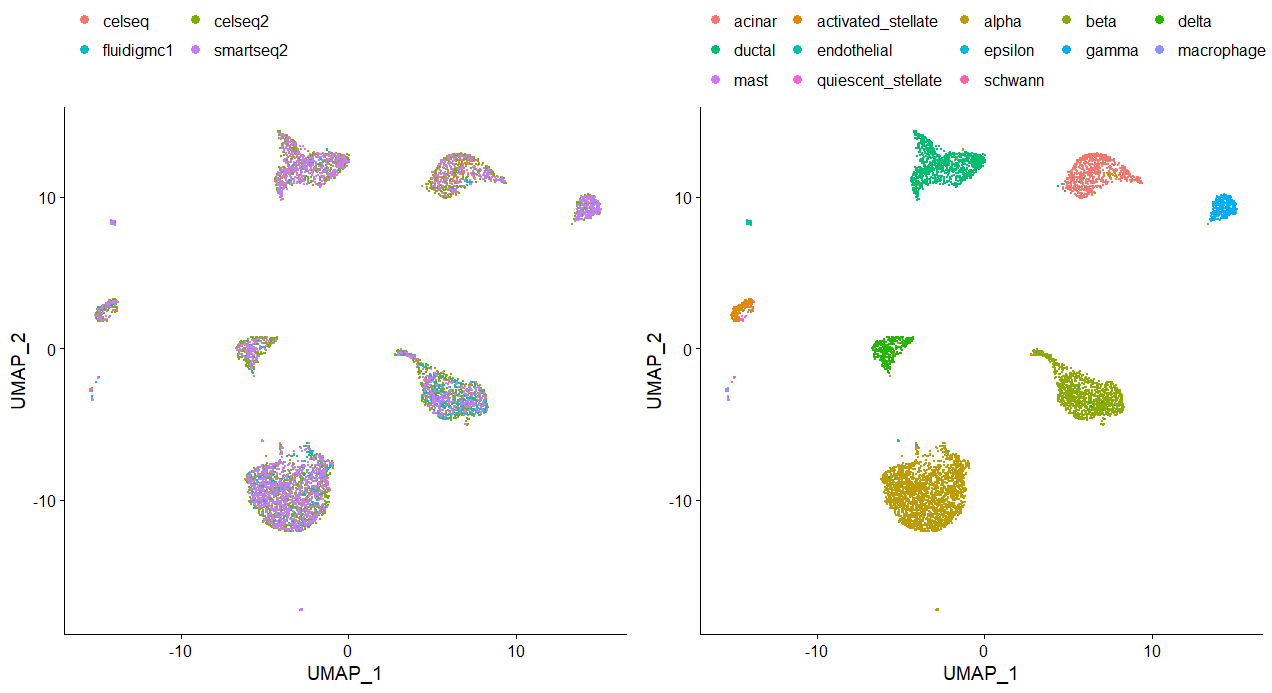
四、使用另一个数据集来验证该流程
安装数据集
InstallData("pbmcsca")
数据预处理
data("pbmcsca")
pbmc.list <- SplitObject(pbmcsca, split.by = "Method")
for (i in names(pbmc.list)) {
pbmc.list[[i]] <- SCTransform(pbmc.list[[i]], verbose = FALSE)
}
pbmc.features <- SelectIntegrationFeatures(object.list = pbmc.list, nfeatures = 3000)
pbmc.list <- PrepSCTIntegration(object.list = pbmc.list, anchor.features = pbmc.features)
pbmc.anchors <- FindIntegrationAnchors(object.list = pbmc.list, normalization.method = "SCT",
anchor.features = pbmc.features)
pbmc.integrated <- IntegrateData(anchorset = pbmc.anchors, normalization.method = "SCT")
pbmc.integrated <- RunPCA(object = pbmc.integrated, verbose = FALSE)
pbmc.integrated <- RunUMAP(object = pbmc.integrated, dims = 1:30)
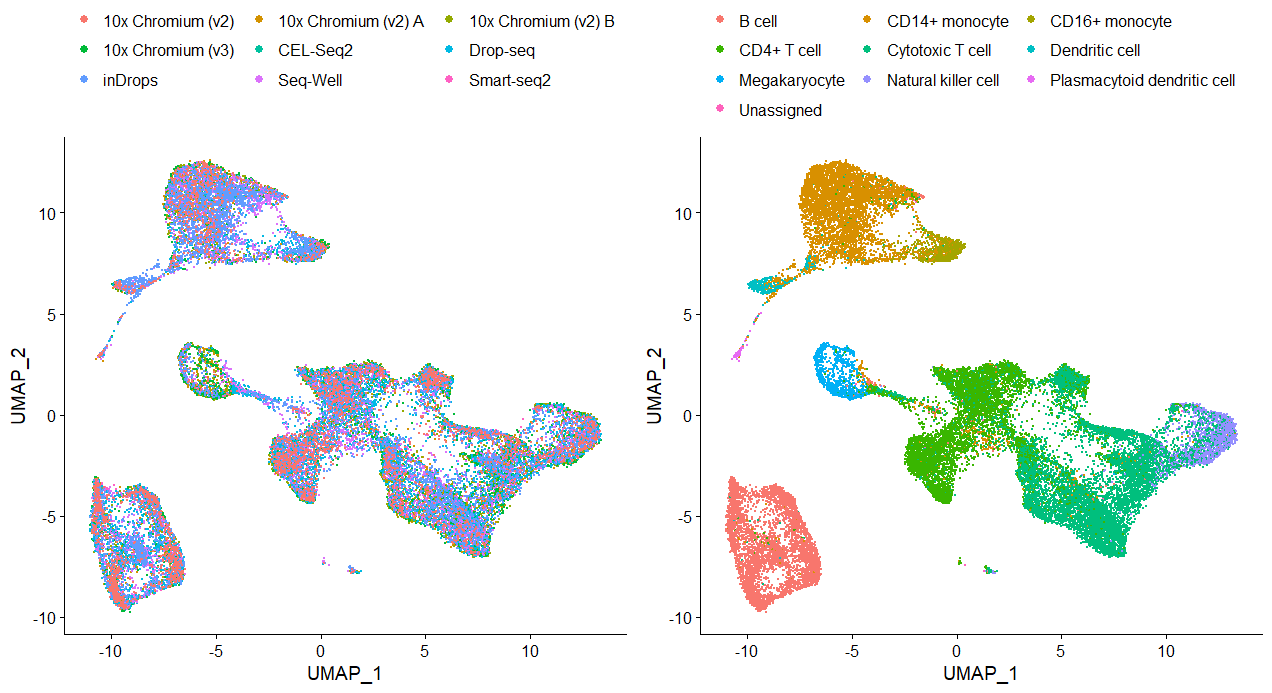
plots <- DimPlot(pbmc.integrated, group.by = c("Method", "CellType"), combine = FALSE)
plots <- lapply(X = plots, FUN = function(x) x + theme(legend.position = "top") + guides(color = guide_legend(nrow = 4,
byrow = TRUE, override.aes = list(size = 2.5))))
CombinePlots(plots)


















 426
426

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











