






基于 RNA 数据的分析还有很多展示形成,我这里都会一次介绍,以及最后的 SCI 文章中的组图,完成所有分析流程,首先讲下 MA 图形的绘制流程,这里还是非常全面的,仅供参考!
MA plot
MA-plot (M-versus-A plot),也称为 Bland-Altman plot,主要应用在基因组数据or 转录组的数据展示,主要是对于数据分布情况的可视化。该图将数据转换为M(对数比)和 A(平均值),然后绘制这些值来可视化两个样本中测量值之间的差异。
原理
早期 MA 图主要应用于 DNA 芯片数据,现在常用于高通量测序数据中基因差异表达分析结果的展示,其公式如下:
M 常对应差异表达分析获得的差异对比组之间基因表达变化 log2FC。A 可以利用差异对比组的 FPKM 进行计算,以 R 和 G 来表示差异对比组的话,可以取 R 组基因的平均 FPKM和 G 组基因的平均 FPKM 进行计算。MA 图一般来说,M 一般做Y轴,A 一般做 X 轴。多个 R 包可以实现 MA 图绘制,如下:
-
affy (ma.plot,mva.pairs)
-
marray (maPlot)
-
edgeR (plotSmear)
-
limma (plotMA)
下面我们就分别介绍这四种方式详细流程,实现 MA 绘图,其中涉及到两套数据一个是芯片 array 的数据,一个是 NGS 数据集,根据自己的数据类型选择对应的软件包来完成绘图。
1. affy (ma.plot,mva.pairs)
affy 软件包是最早分析 Affymetrix GeneChip expression 的数据开发的,之后很长一段时间引用量还是蛮高的,随着 NGS 的出现,大家都会更倾向使用 DESeq 等针对高通量测序的软件包。我们以软件包里面的数据为例子,讲解一下 MA 图的绘制。
软件包安装包括 affy 和 数据来源软件包 affydata,如下:
if (!require("BiocManager", quietly = TRUE))
install.packages("BiocManager")
if (!require("affy", quietly = TRUE))
BiocManager::install("affy")
if (!require("affy", quietly = TRUE))
BiocManager::install("affydata")
加载 affy,affydata 软件包,如下:
library(affy)
library(affydata)
数据调取 Dilution 来自 affydata 软件包,数据说明,如下:
Two sources of cRNA A (human liver tissue) and B (Central Nervous System cell line) have been hybridized to human array (HGU95A) in a range of proportions and dilutions. This data set is taken from arrays hybridized to source A at 10.0 and 20 μg. We have two replicate arrays for each generated cRNA. Three scanners have been used in this study. Each array replicate was processed in a different scanner.
data(Dilution)
print(Dilution)
## AffyBatch object
## size of arrays=640x640 features (38422 kb)
## cdf=HG_U95Av2 (12625 affyids)
## number of samples=4
## number of genes=12625
## annotation=hgu95av2
## notes=
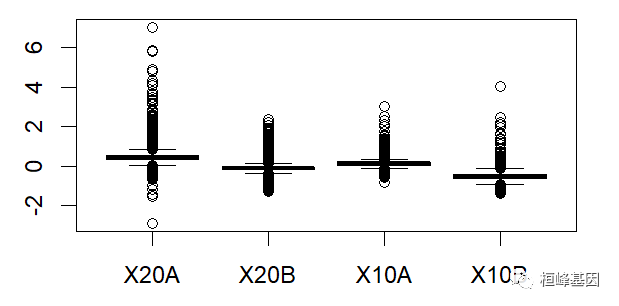
数据中包括四个样本,那么对单个样本绘制 MA 图,其中 IQR(interquartile range) 指四分位距又称四分差。是描述统计学中的一种方法,以确定第三四分位数和第一四分位数的区别。与方差、标准差一样,表示统计资料中各变量分散情形,但四分差更多为一种稳健统计(robust statistic)。
MAplot(Dilution)
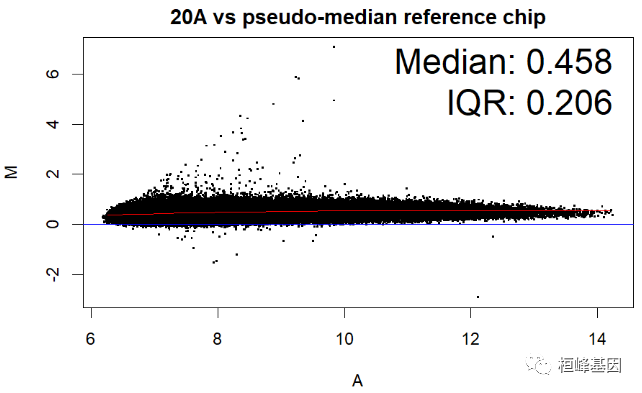
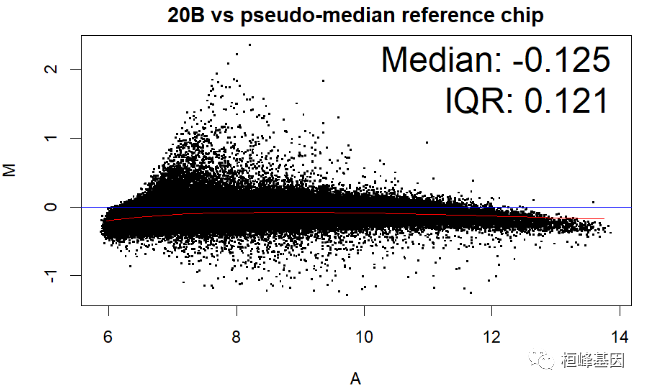
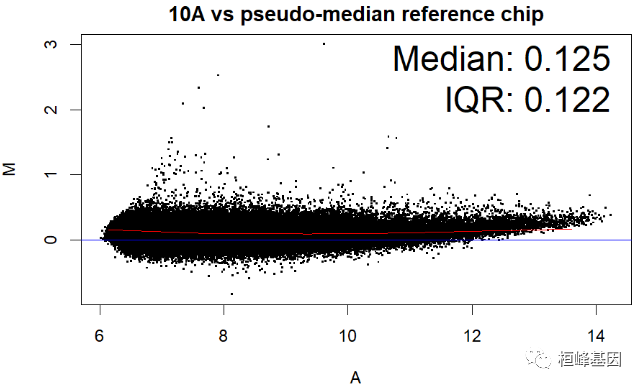
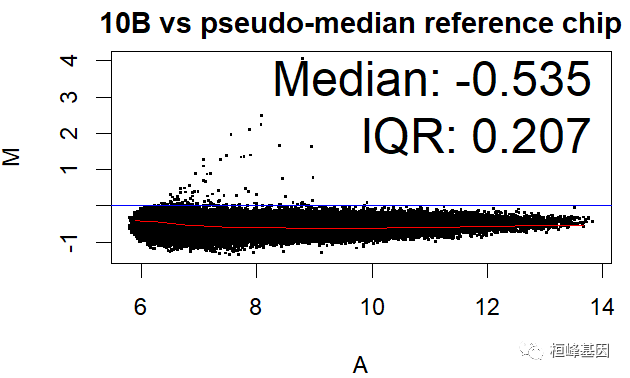
绘制箱式图再次看下四个样本的表达数据分布,如下:
Mbox(Dilution)
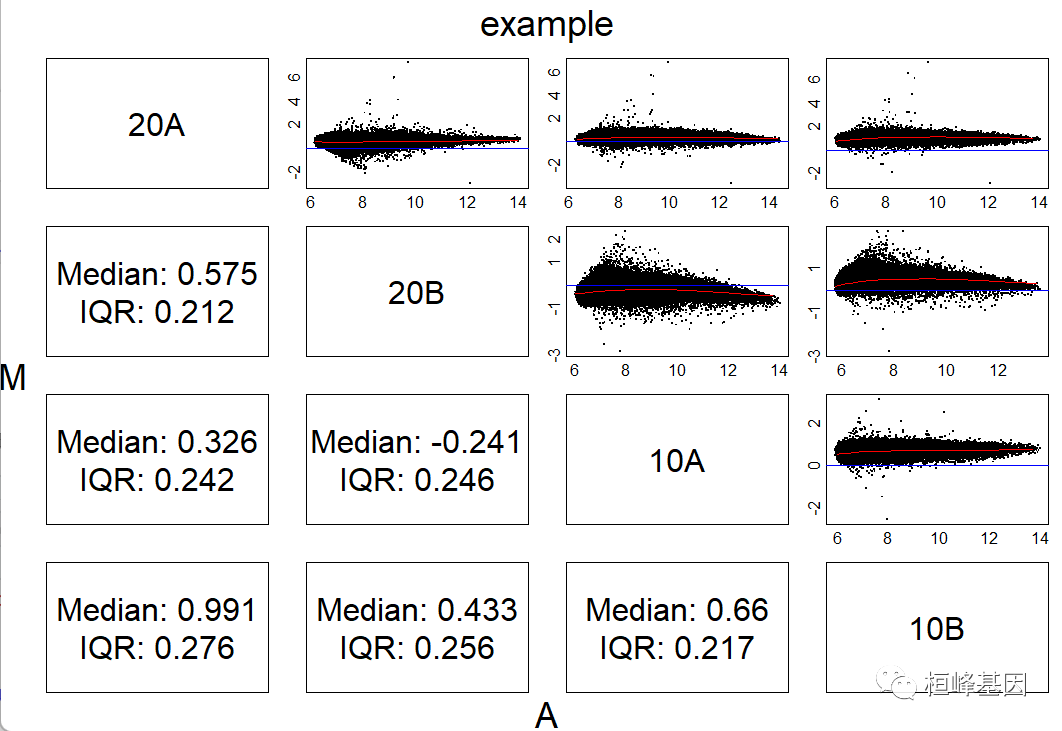
四个样本两两比较,如下:
mat<-pm(Dilution) ## get indexes for the PM probes
mva.pairs(mat,log=TRUE,main="example")
2. marray (maPlot)
看下marray 软件包里面的例子,这个软件包同样也是针对基因芯片数据来绘制 MA 图形。
软件安装及加载,如下:
if (!require("marray", quietly = TRUE))
BiocManager::install("marray")
library(marray)
数据调取 swirl 来自 marray 软件包的数据,是 Gene expression data from Swirl zebrafish cDNA microarray experiment. The swirlRaw dataset consists of an object swirl of class marrayRaw, which represents pre-normalization intensity data for a batch of cDNA microarrays.
# Examples use swirl dataset, for description type ? swirl
data(swirl)
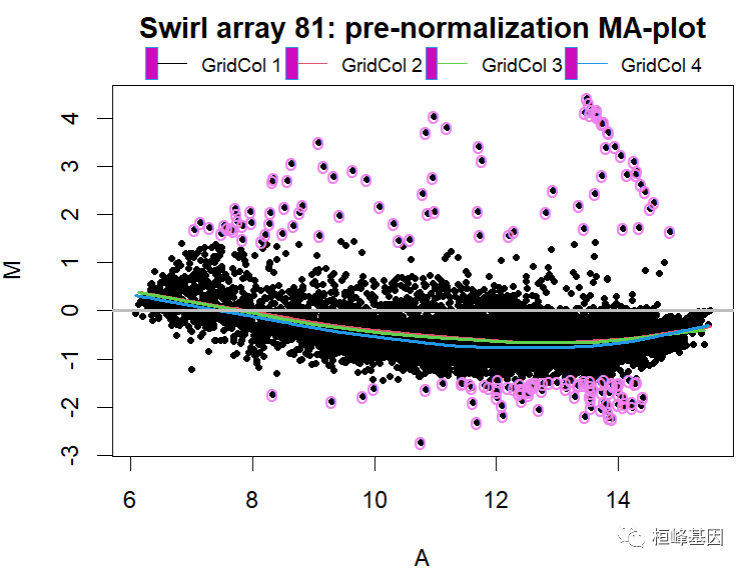
绘制 MA 图,如下:
# Pre-normalization MA-plot for the Swirl 81 array, with the lowess fits for # individual grid columns and 1% tails of M highlighteddefs <- maDefaultPar(swirl[, 1], x = "maA", y = "maM", z = "maGridCol")
defs$def.legend$x=6defs$def.legend$y=6.5defs$def.legend$bty="n"defs$def.legend$lwd=1defs$def.legend$horiz=TRUEdefs$def.legend$xpd=TRUEdefs$def.legend$cex=0.8
legend.func <- do.call("maLegendLines", defs$def.legend)lines.func <- do.call("maLowessLines", c(list(TRUE, f = 0.3), defs$def.lines))text.func<-maText(subset=maTop(maM(swirl)[,1],h=0.01,l=0.01), labels="o", col="violet")maPlot(swirl[, 1], x = "maA", y = "maM", z = "maGridCol", lines.func=lines.func, text.func = text.func, legend.func=legend.func, main = "Swirl array 81: pre-normalization MA-plot")
3. edgeR (plotSmear)
软件安装及加载,如下:
if (!require("edgeR", quietly = TRUE))
BiocManager::install("edgeR")
library(edgeR)
数据读取,我们仍然使用 TCGA-COAD 数据集的表达数据,数据文件的读取处理等都是之前讲过,这里为了保证流程的完整性,同样给出来,如下:
if (!requireNamespace("BiocManager", quietly = TRUE))
BiocManager::install("BiocManager")
if (!requireNamespace("edgeR", quietly = TRUE))
BiocManager::install("edgeR")
## 数据文件读取
library(TCGAbiolinks)
# 请求数据。
query <- GDCquery(project ="TCGA-COAD",
data.category = "Transcriptome Profiling",
data.type ="Gene Expression Quantification" ,
workflow.type="HTSeq - Counts")
samplesDown <- getResults(query,cols=c("cases"))
dataSmTP <- TCGAquery_SampleTypes(barcode = samplesDown,
typesample = "TP")
# 从samplesDown中筛选出NT(正常组织)样本的barcode
#此处共检索出50个NT样本barcodes
dataSmNT <- TCGAquery_SampleTypes(barcode = samplesDown,
typesample = "NT")
group<-as.data.frame(list(Sample=c(dataSmTP,dataSmNT),
Group=c(rep("TP",length(dataSmTP)),rep("NT",length(dataSmNT)))))
###设置barcodes参数,筛选符合要求的371个肿瘤样本数据和50正常组织数据
queryDown <- GDCquery(project = "TCGA-COAD",
data.category = "Transcriptome Profiling",
data.type = "Gene Expression Quantification",
workflow.type = "HTSeq - Counts",
barcode = c(dataSmTP, dataSmNT))
#GDCdownload(queryDown,
# method = "api",
# directory = "GDCdata",
# files.per.chunk = 10)
dataPrep1 <- GDCprepare(query = queryDown,
save = TRUE,
save.filename = "COAD_case.rda")
dataPrep2 <- TCGAanalyze_Preprocessing(object = dataPrep1,
cor.cut = 0.6,
datatype = "HTSeq - Counts")
dim(dataPrep2)
table(group$Group)
利用 DESeq2 做差异分析,这里就是用 DESeq2 差异基因结果举例子,如下:
#(1)构建 DGEList 对象
dgelist <- DGEList(counts = dataPrep2, group = group$Group)
#(2)过滤 low count 数据,例如 CPM 标准化(推荐)
keep <- rowSums(cpm(dgelist) > 1 ) >= 2
dgelist <- dgelist[keep, , keep.lib.sizes = FALSE]
#(3)标准化,以 TMM 标准化为例
dgelist_norm <- calcNormFactors(dgelist, method = 'TMM')
##04. 差异表达分析
#差异表达基因分析
#首先根据分组信息构建试验设计矩阵,分组信息中一定要是对照组在前,处理组在后
design <- model.matrix(~group$Group)
#(1)估算基因表达值的离散度
#install.packages("statmod")
library(statmod)
dge <- estimateDisp(dgelist_norm, design, robust = TRUE)
#(2)模型拟合,edgeR 提供了多种拟合算法
#负二项广义对数线性模型
fit <- glmFit(dge, design, robust = TRUE)
lrt <- glmLRT(fit)
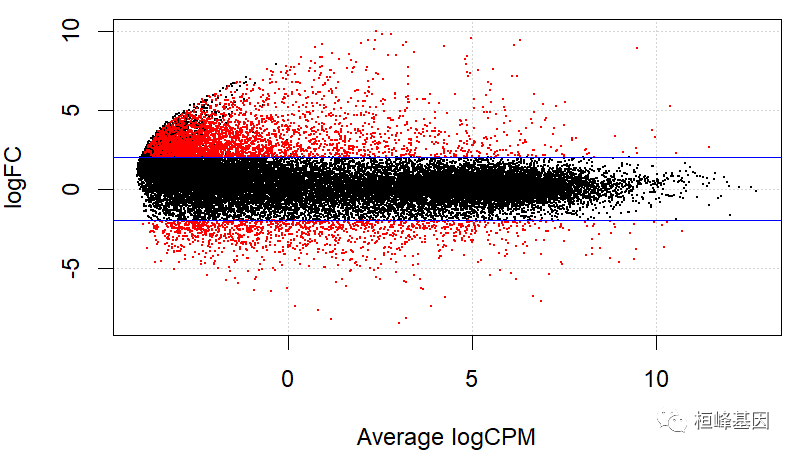
利用上面的结果绘制 MA 图,这里面绘图函数plotSmear 是 edgeR 软件包内置的函数,所以在整个软件包计算出来的差异表达结果可直接读入该函数,我们选择pvalue < 0.001, foldchange = 2,获得上调基因 3230 下调基因 1298,其他为不显著基因,如下:
deGenes <- decideTestsDGE(lrt, p=0.001, lfc = 2)
summary(deGenes)
## group$GroupTP
## Down 1298
## NotSig 27249
## Up 3230
detag <- rownames(lrt)[as.logical(deGenes)]
利用差异表达结果,非常方便即可完成绘图,如下:
plotSmear(lrt, de.tags=detag)
abline(h=c(-2, 2), col='blue')
4. limma (plotMA)
软件包 limma 中 函数plotMA 同样也可以实现 MA 图的绘制,同样也可以针对基因芯片数据来绘制 MA 图形,NGS也可以使用。
软件安装及加载,如下:
if (!require("limma", quietly = TRUE))
BiocManager::install("limma")
library(limma)
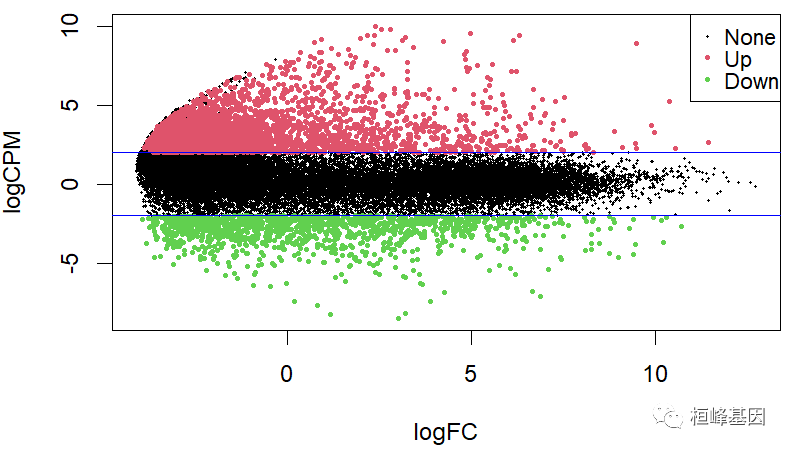
我们选择pvalue < 0.001, foldchange = 2 的cutoff,获得上调基因 3230 下调基因 1298,其他为不显著基因。M 常对应差异表达分析获得的差异对比组之间基因表达变化 log2FC,而 A 对应表达的平均数 logCPM,绘制 MA 图,如下:
res <- topTags(lrt, n = nrow(dgelist$counts))
res<-as.data.frame(res)
res <- res[order(res$FDR, res$logFC, decreasing = c(FALSE, TRUE)), ]
res[which(res$logFC >= 2 & res$FDR < 0.001),'sig'] <- 'Up'
res[which(res$logFC <= -2 & res$FDR < 0.001),'sig'] <- 'Down'
res[which(abs(res$logFC) <= 2 | res$FDR >= 0.001),'sig'] <- 'None'
MA <- new("MAList")
MA$M = res$logFC
MA$A = res$logCPM
status=res$sig
plotMA(MA,status = status,xlab = "logFC",ylab = "logCPM",hl.cex = 0.5)
abline(h=c(-2, 2), col='blue')
此次 MA 绘图完成教程整理的非常全面,大家一定要关注公众号,后期会出视频版和直播课程,敬请关注!

桓峰基因
生物信息分析,SCI文章撰写及生物信息基础知识学习:R语言学习,perl基础编程,linux系统命令,Python遇见更好的你
41篇原创内容
公众号
Reference:
-
Gautier, L., Cope, L., Bolstad, B. M., and Irizarry, R. A. 2004. affy—analysis of Affymetrix GeneChip data at the probe level. Bioinformatics 20, 3 (Feb. 2004), 307-315.
-
S. Dudoit and Y. H. Yang. (2002). Bioconductor R packages for exploratory analysis and normalization of cDNA microarray data. In G. Parmigiani, E. S. Garrett, R. A. Irizarry and S. L. Zeger, editors, The Analysis of Gene Expression Data: Methods and Software, Springer, New York.
-
Ritchie, ME, Phipson, B, Wu, D, Hu, Y, Law, CW, Shi, W, and Smyth, GK (2015). limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Research Volume 43, e47.
-
Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26(1):139-140

















 582
582

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









