







从头学习生信:感谢建明老师教程:
阅读文献并下载原始测序数据之helicos转录组数据 | 生信菜鸟团 (bio-info-trainee.com)
阅读文献并下载原始测序数据之helicos转录组数据
目录
- 阅读pdf文献,并找到原始数据搜索关键词。
- 根据关键词在NCBI的SRA板块搜索找到其下载地址
- 根据下载地址写批处理批量下载所有原始测序数据
- 用NCBI提供的工具解压SRR数据,还原成fastq格式reads
正文
一、阅读pdf文献,并找到原始数据搜索关键词

可以看到它的下载索引是SRP003040,阅读文献可知其包含4种细胞的6种处理方式的转录组数据
二、根据关键词在NCBI的SRA板块搜索找到其下载地址
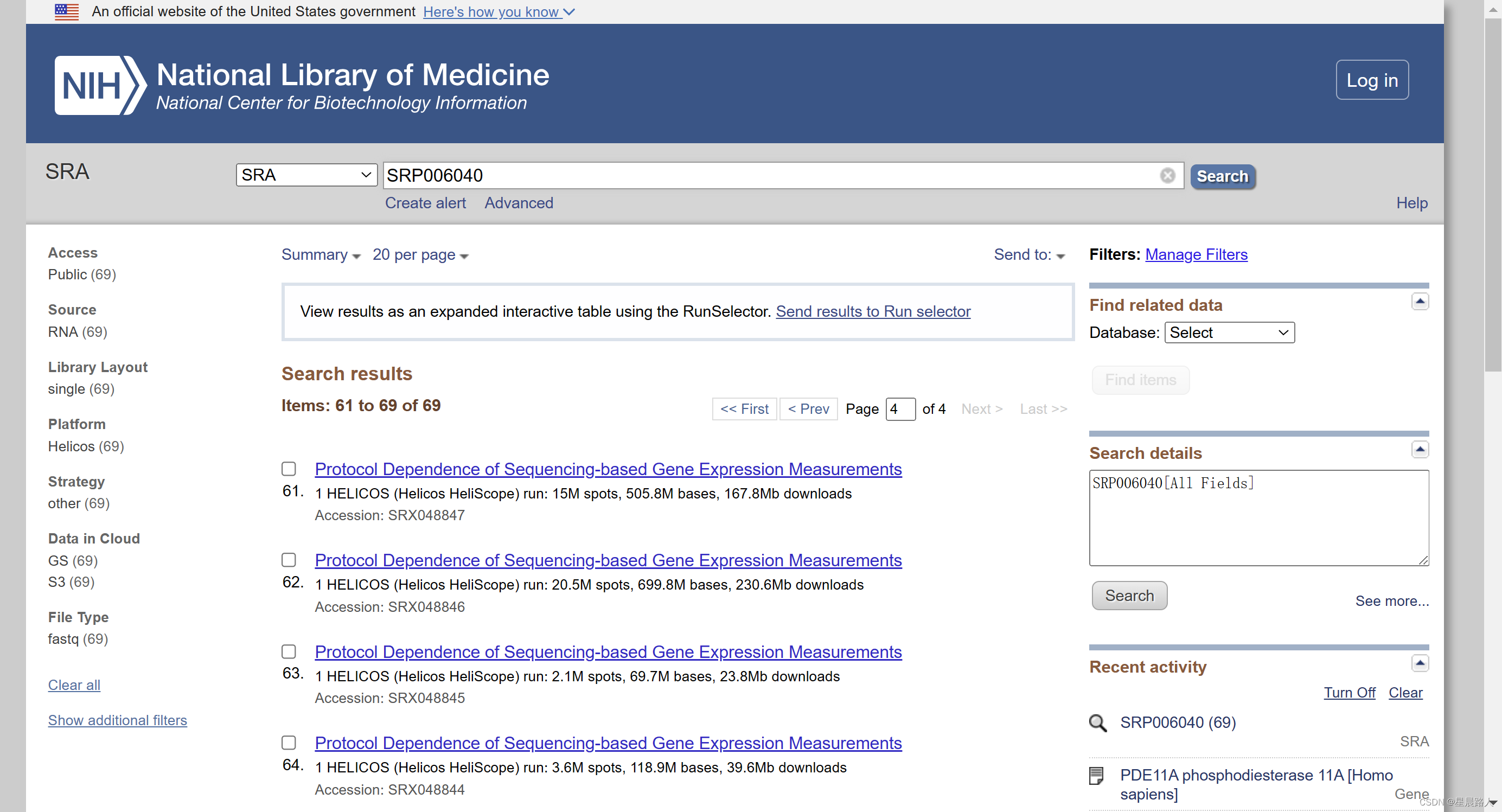 三、 根据下载地址写批处理批量下载所有原始测序数据
三、 根据下载地址写批处理批量下载所有原始测序数据
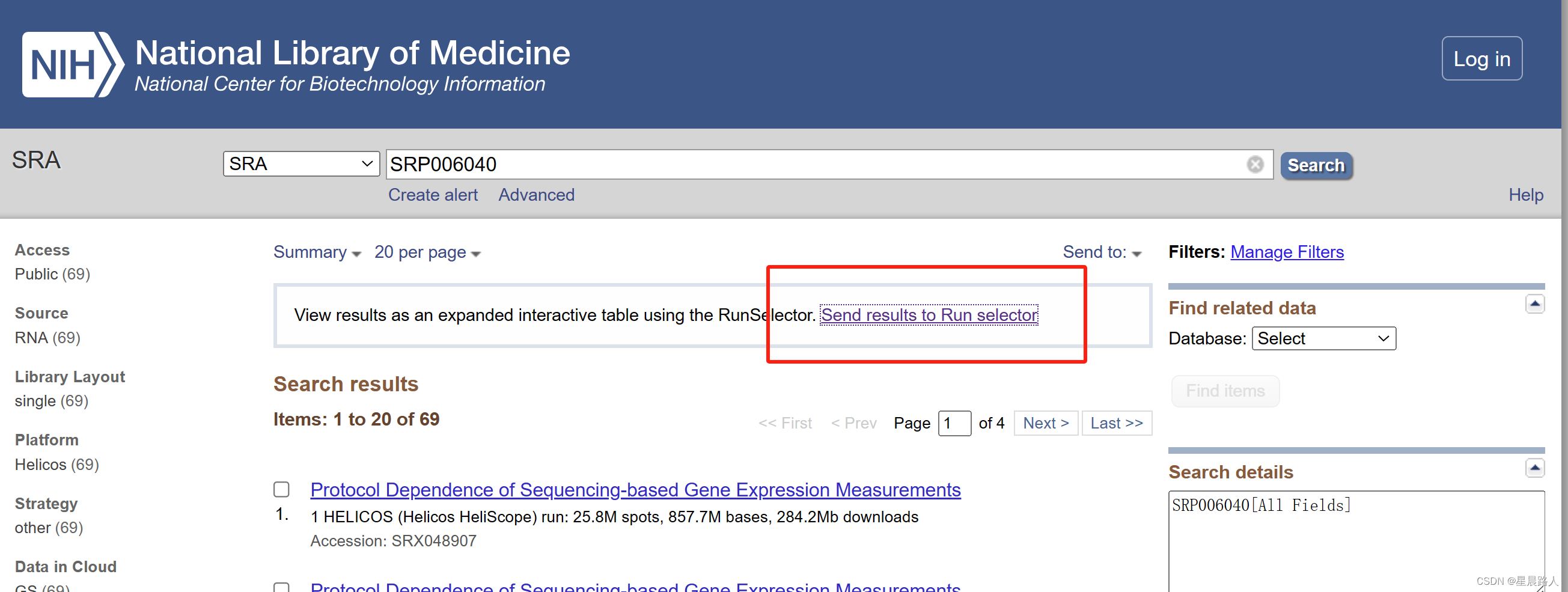
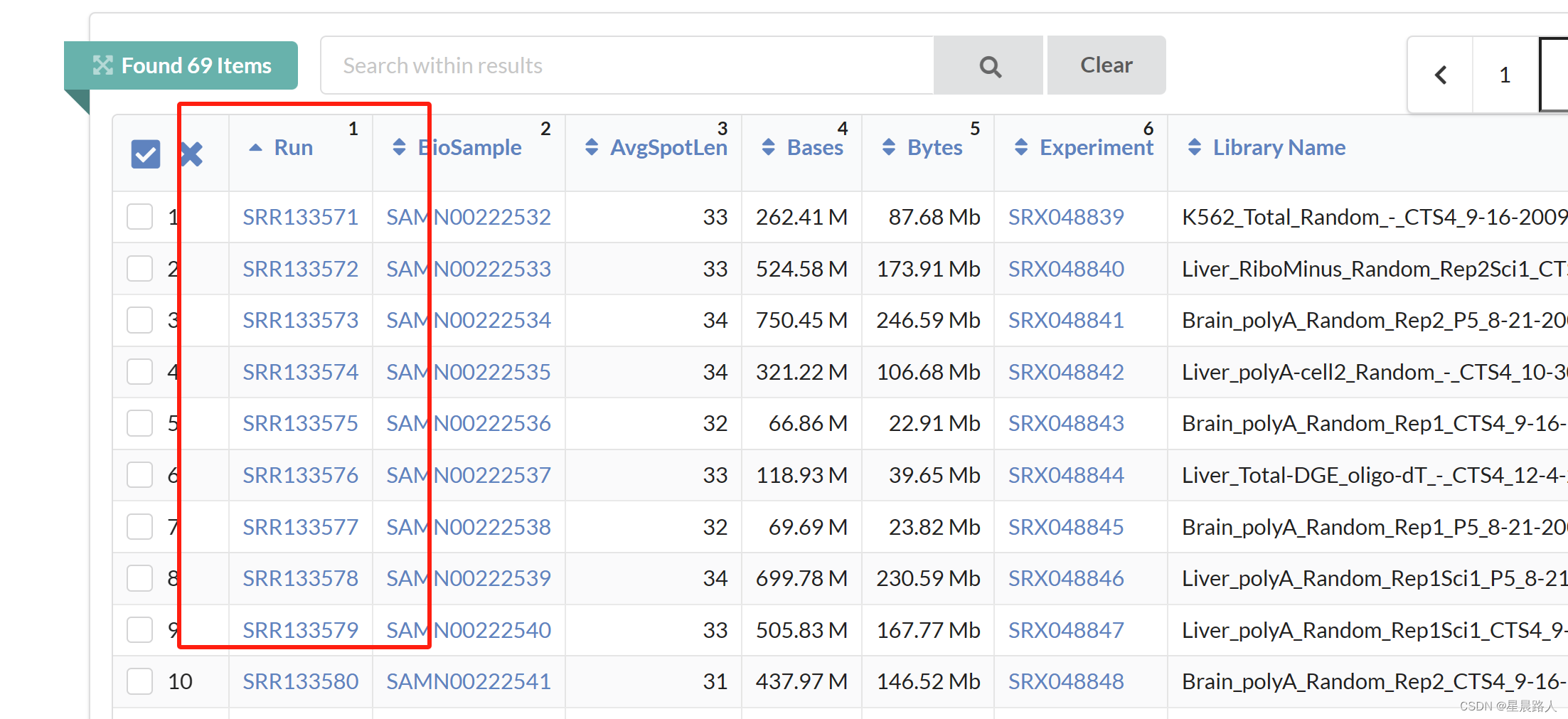
解析SRA地址可知从SRR133571.sra到SRR133639.sra,共69个文件
将SRR整理到文件id中
批处理代码如下:
- while read id
- do
- echo $id
- wget ftp://ftp-trace.ncbi.nlm.nih.gov/sra/sra-instant/reads/ByRun/sra/SRR/SRR133/$id/$id.sra
- done <$1
下载之后共14G的数据
四、用NCBI提供的工具解压SRR数据,还原成fastq格式reads
也是批处理进行解压,代码如下
- for i in *sra
- do
- /home/jmzeng/bio-soft/sratoolkit.2.3.5-2-ubuntu64/bin/fastq-dump --split-3 $i
- Done
解压后共216G的数据,都是fastq格式的单端50bp的数据。
This entry was posted in 基础数据库 by ulwvfje. Bookmark the permalink.

















 3122
3122

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









