






本文版权归《遗传》杂志,已获授权,转载请联系杂志社

微生物组数据分析方法与应用
刘永鑫1,2,秦媛1,2,3,郭晓璇1,2,白洋1,2,3
1. 中国科学院遗传与发育生物学研究所,植物基因组学国家重点实验室,北京 100101
2. 中国科学院遗传与发育生物学研究所,中国科学院–英国约翰英纳斯中心植物和微生物科学联合研究中心,
北京 100101
3. 中国科学院大学现代农学院,北京 100049
原文链接:http://www.chinagene.cn/CN/10.16288/j.yczz.19-222
录用日期:2019-09-02
引文:刘永鑫, 秦媛, 郭晓璇 & 白洋. 微生物组数据分析方法与应用. 遗传. 2019, 41: 1-18. doi:10.16288/j.yczz.19-222
Yong-Xin Liu, Yuan Qin, Xiaoxuan Guo & Yang Bai. Methods and applications for microbiome data analysis. Hereditas(Beijing). 2019, 41: 1-18. doi:10.16288/j.yczz.19-222
有声版正文,校稿阶段录音,41分钟。
摘要: 高通量测序技术的发展衍生出一系列微生物组(microbiome)研究技术,如扩增子、宏基因组、宏转录组等,快速推动了微生物组领域的发展。微生物组数据分析涉及的基础知识、软件和数据库较多,对于同领域研究者开展学习和选择合适的分析方法具有一定困难。本文系统概述了微生物组数据分析的基本思想和基础知识,详细总结比较了扩增子和宏基因组分析中的常用软件和数据库,并对高通量数据下游分析中常用的几种方法,包括统计和可视化、网络分析、进化分析、机器学习和关联分析等,从可用性、软件选择以及应用等几个方面进行了概述。本文拟通过对当前微生物组主流分析方法的整理和总结,为同领域研究者更方便、灵活的开展数据分析,快速选择研究分析工具,高效挖掘数据背后的生物学意义提供参考,进一步推动微生物组研究在生物学领域的发展。
关键词: 微生物组;数据分析;扩增子;宏基因组;分析流程
Methods and applications for microbiome data analysis
Yong-Xin Liu1,2, Yuan Qin1,2,3, Xiaoxuan Guo1,2, Yang Bai1,2,3
1. State Key Laboratory of Plant Genomics, Institute of Genetics and Developmental Biology, The Innovative Academy of Seed Design, Chinese Academy of Sciences, Beijing 100101, China
2. CAS-JIC Centre of Excellence for Plant and Microbial Science, Institute of Genetics and Developmental Biology, Chinese Academy of Sciences, Beijing 100101, China
3. College of Advanced Agricultural Sciences, University of Chinese Academy of Sciences, Beijing 100101, China
Abstract: Development of high-throughput sequencing stimulates a series of microbiome technologies, such as amplicon sequencing, metagenome, metatranscriptome, which have rapidly promoted microbiome research. Microbiome data analysis involves a lot of basic knowledge, softwares and databases, and it is difficult for peers to learn and select proper methods. This review systematically outlines the basic ideas of microbiome data analysis and the basic knowledge required to conduct analysis. In addition, it summarizes the advantages and disadvantages of commonly used softwares and databases used in the comparison, visualization, network, evolution, machine learning and association analysis. This review aims to provide a convenient and flexible guide for selecting analytical tools and suitable databases for mining the biological significance of microbiome data.
Keywords: microbiome; data analysis; amplicon; metagenome; pipeline
微生物组(microbiome)是指包括细菌、古菌、低(高)等真核生物、病毒等微生物的基因和基因组,及其周围环境在内的全部[1]。研究表明微生物组在人类和动植物的营养吸收[2]、疾病抵抗[3]和环境适应中起重要作用[4,5]。
近年来第二代测序(next generation sequencing, NGS)技术的发展使得基于非培养方法研究微生物组成为可能,并推动了微生物组研究进入了黄金发展时期[6]。目前对微生物组样本的研究主要集中在3个层面(图1A):(1)微生物培养层面:培养组学(Culturome)是该层面最重要的研究手段。通过在固体培养皿挑单菌落或使用96孔板液体高通量培养的方式获得微生物群落中可培养的菌落,随后结合标记基因(marker gene)测序、分离纯化等方法进行菌种鉴定和保藏。目前该方法已在人类[7]、拟南芥(Arabidopsis thaliana)[8]、水稻(Oryza sativa)[9]等物种中应用和报道;(2) DNA层面:针对DNA易于提取和保存的特点,研究者相继发展出扩增子(amplicon)、宏基因组(metagenome)[10]和宏病毒组(metavirome)等测序研究手段[11]。扩增子测序常用的标记基因主要包括原核生物的16S rRNA基因、真核生物的18S rRNA基因以及转录间隔区(internal transcribed spacers, ITS)等。由于扩增子测序仅能获得研究对象的物种组成信息,要想进一步研究物种所携带的其他功能基因,就需要开展宏基因组测序和分析;(3) mRNA层面:通过对微生物组样本提取RNA进行宏转录组(metatranscriptome)测序,可以根据微生物组样本中的基因表达谱进一步揭示微生物群落原位功能[12]。病毒包括DNA和RNA病毒两大类,想要全面开展宏病毒组学研究需要宏基因组结合宏转录组测序(图1A)。
鉴于微生物组编码的基因近千万[13],想要从微生物组海量数据中挖掘有效信息,必须了解和掌握本领域相关软件和数据库的使用,才能在计算机或服务器上开展可重现(reproducible)的数据分析。而传统的生物学家由于生物信息学知识相对薄弱、微生物组数据分析经验不足等情况,在数据分析过程中经常会面临Linux使用、代码重用和软件选择等众多困难。本文系统概述了当前微生物组数据主流分析的基本思路和步骤,同时对开展微生物组数据分析提供了建议,最后对本领域常用分析方法的优缺点和适用范围进行总结,以期对同行更高效地开展微生物组数据分析,挖掘大数据背后的生物学规律有所帮助。
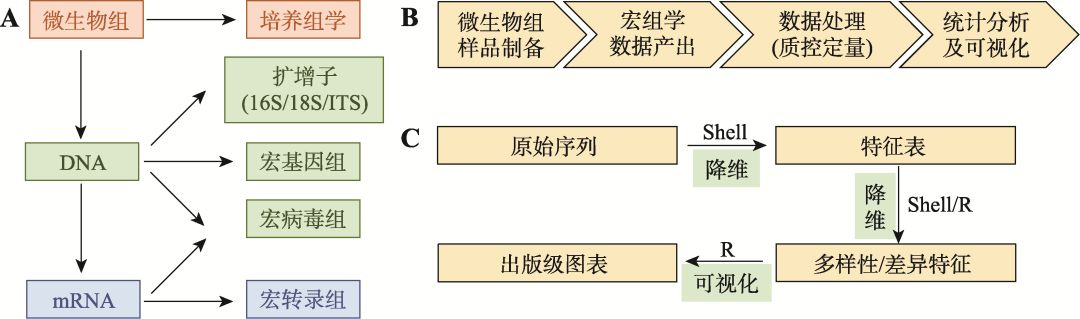
图1 微生物组研究方法概述
Fig. 1 Methods in microbiome research
A:微生物组常用的研究层面和对应方法。微生物组按研究层面主要分为微生物培养、DNA和mRNA等3个层面;按研究技术主要包括培养组学(culturome)、扩增子(amplicon)、宏基因组(metagenome)、宏病毒组(metavirome) 和宏转录组(metatranscriptome)等测序技术[1,12]。B:微生物组研究的基本步骤。基于测序技术为基础的微生物组研究,主要分为样本制备、测序、数据处理和统计分析四个阶段。C:微生物组数据分析的基本步骤、常用环境和思想。组学数据分析主要分3步,图中箭头上描述了实现分析的常用语言环境Shell和/或R;图中箭头下展示各步分析的目的,即通过降维和可视化的基本思想,实现将大数据转化为可读图表。
1 微生物组数据分析的基本步骤
微生物组研究主要分为4个阶段(图1B):(1)微生物组样品制备:基于科学的实验设计,采集来自人、动植物或环境中的微生物组样本,并根据研究的目的,选择提取DNA或RNA等;(2)宏组学(meta- omics)数据产出:抽提样品的DNA或RNA后,通过构建测序文库和进行高通量测序来获得宏组学数据。例如,扩增子16S rRNA基因片段主要采用双端250 bp (pair-end 250 bp, PE250)测序,单个样本3~5万条序列的深度;宏基因组多采用PE150测序,获得微生物部分至少2千万条序列(150 bp220 Mb = 6 Gb);(3)数据处理(质控定量):当获得微生物组数据后,首先要进行质量控制,包括去除测序和建库过程中人为添加的引物、接头以及测序过程中产生的低质量序列等。此外,宿主相关的微生物组测序结果中含有大量宿主序列,需采用比对宿主基因组的方式去除。获得的纯净序列(clean data)再比对至参考数据库或从头(De novo)组装的参考基因集,定量为特征表(feature table),根据序列注释类型可将特征表分为物种或功能基因组成表;(4)统计分析和可视化:特征表还需要进一步结合样本元数据(metadata)进行统计分析,并选择合适的图形进行可视化,有利于生物学规律的观察和总结,提高结果的可读性和传播性(图1B)。本文将主要对第3和第4步骤做进一步讨论和总结。
当获得微生物组原始数据后,如何对其进行分析至高可读性的出版级别图表?为便于理解,本文将微生物组数据分析过程划分为3个主要步骤(图1C):
第一步:原始数据转换为特征表。微生物组数据通常为NGS产生的fastq格式序列文件,包括碱基序列和质量值,序列数量级可达106~109条。这就需要在高效的Shell环境下使用命令行工具对大数据进行质控和定量,降维至数量级为103~105的特征表。特征表常为计数型数据(count data),如物种分类学(taxonomy)表、可操作分类单元(operational taxonomic unit, OTU)表、扩增序列变异(amplicon sequence variant, ASV)表、基因丰度(gene abundance)表和通路丰度(pathway abundance)表等。
第二步:特征表转换为多样性和/或差异特征。例如,微生物组研究中扩增序列变异表和基因丰度表仍然很大,因此研究者常采用Alpha或Beta多样性分析、物种或功能层级注释、差异比较等方法,将数据表进一步降维至101~103。该数据结果更方便研究者运用专业知识挖掘规律和解释生物学问题。
第三步:数据可视化为出版级图表。近年来可视化语言和工具的发展提高了数据挖掘和结果解读的效率,如折线柱、柱状图、箱线图、散点图和热图等的广泛使用,更易于帮助研究者发现数据中的规律(图1C)。
从微生物组数据分析的全过程中可以看出,降维和可视化是大数据分析的核心指导思想,即把数据降维至可读的数量,通过可视化分析方便同领域研究者阅读和传播。实现这两个过程主要涉及两种语言环境,即首先通过Linux系统中的Shell语言配合工具软件实现大数据分析和降维,然后利用R语言(https://www.r-project.org)实现基于特征表的统计和可视化。因此熟悉Shell和R这两门语言的基础操作即可满足研究者微生物组数据分析的绝大多数需求。当然,微生物组分析中也常涉及Perl、Python、Java等语言的使用,它们更多作为软件和脚本在Shell环境下运行,用户可以根据自己的基础和习惯选择不同的语言环境进行分析和可视化。
2 微生物组数据分析常用的环境
微生物组数据分析需要在专门的语言环境下开展,熟悉常用的语言环境能够帮助我们更好地利用现有工具开展数据分析。目前本领域的分析工具主要集中在Shell和R两种语言环境下运行。几乎所有的服务器都是Linux系统,默认的Shell环境自带上百个命令和Bioconda近万个生物信息软件可快速搭建各种分析流程[14]。R语言开源免费,官网CRAN (https://cran.r-project.org/)发布了14767个统计和可视化包,Bioconductor (http://www.bioconductor.org)上更有1741个生物学专用包(数量统计截止2019年8月20日),可实现最灵活的统计分析。掌握这两门语言基础,可以高效地利用现有软件开展数据分析、统计和可视化。本文重点介绍Shell和R语言,是因为这两类语言环境下有非常多可利用的生物学软件(包),用户可以通过极少的代码串联现有工具来实现数据分析。特别是对于初级使用者来说,学习和应用相对更加便捷。
Shell语言是与Linux系统交互命令的合集,几乎所有的微生物组分析工具都有可以在Linux服务器的的Shell环境下运行,而在其他环境中搭建分析流程非常困难。如果用户的电脑为Windows系列,需要安装远程访问Linux服务器的软件,如XShell、putty或ssh secure shell等,这里推荐使用商业化开发且对学校免费的XShell。而Mac系统是类UNIX系统内核,系统自带的Terminal程序即可实现远程访问Linux。R语言自带图形界面RGui,可以实现交互式统计分析和可视化,RScript命令可在命令行下执行R脚本。近两年快速发展的集成开发环境RStudio (https://www.rstudio.com/),自2018年升级至1.1版后同时支持Shell和R脚本的编辑和运行。RStudio是跨平台软件,在Windows/ Mac/Linux上都可以轻松安装,还有服务器版本可以在网页中运行,保证不同终端无需安装任何额外程序,即可保持数据分析工作环境的一致性。对于初学数据分析的研究者来说,可通过学习RStudio来掌握数据分析、代码管理、程序调试、结果图片调整和保存等操作。
有了好用的分析代码管理工具,还需要学习语言基础读懂分析代码,才能使用和修改现有的分析流程和方法。对于以数据分析为主的研究者,建议系统学习Shell和R语言基础。Shell语言推荐学习《鸟哥的LINUX私房菜基础学习篇(第四版)》,其中Linux的基本命令、文件系统和Shell脚本编写可重点学习,服务器管理员还需要学习系统和用户管理等内容。R语言推荐学习《ggplot2:数据分析与图形艺术(第2版)[15],该工具书对系统认识各种图形、了解绘图原理和实现数据可视化非常有帮助。此外,通过学习网络上相关研究者整理总结的的基础知识和代码注释,对于初学者以及偶尔使用数据分析的研究者来说,可能更具有针对性和时效性。
3 微生物组领域常用软件
近10年,随着高通量测序技术的发展和应用,微生物组研究领域的相关分析方法和工具也取得了快速发展,大量优秀的软件、流程和可视化工具相继发布,进一步推动了本领域的发展。
3.1 扩增子分析软件
扩增子分析是微生物组领域应用最广泛的技术,可以快速获悉研究对象中的微生物多样性。本文将重点介绍3款(mothur, QIIME和USEARCH)在近10年内发表且引用过万次的扩增子分析软件(图2),其他更多相关软件介绍详见表1。
(1) Mothur:由美国密歇根大学的Patrick D. Schloss教授团队在2009年发布的首个扩增子分析流程[16]。它整合了之前发表的OTU定义软件DOTUR[17]、OTU差异比较工具SONS[18]以及其他可用工具,实现了第一套较完整的分析流程,让广大研究者开展扩增子分析成为可能(图2)。
(2) QIIME:2010年,美国科罗拉多大学的Rob Knight教授(现单位美国加州大学圣地亚哥分校)团队发布QIIME (发音同chime)分析流程[19]。该流程可在Linux或Mac系统中运行,相比mothur具有更多的优点,主要包括:整合了200多款相关软件和包,实现每个步骤更多软件和方法的选择;提供150多个脚本,实现各种个性化分析,并可以应对不同类型数据和实验设计;流程开放程度高,容易整合新软件和方法;增强统计和可视化,实现多样性、物种组成、差异比较和网络等众多方法和出版级图表绘制。由于QIIME允许同领域研究者较自主地开展扩增子数据的个性化分析和可视化,逐渐成为本领域最受欢迎的软件(图2)。为了满足日益增长的测序数据量和可重复计算的要求,Gregory J. Caporaso教授于2016年起发起了基于Python 3语言从头编写的QIIME 2项目[20]。该项目实现了分析流程的可追溯以满足科研可重复计算的要求;同时推出了一系列新算法,如基于进化距离的快速算法条型(Striped) UniFrac[21]、物种分类新方法q2-feature-classifier[22]等;更重要的是软件的可扩展性和得到了同际同行的广泛支持,如接头和引物序列去除工具cutadapt[23]、序列质量控制R包DADA2[24]、聚类和去冗余的软件VSEARCH[25]、纵向和成对样本分析工具longitudinal[26]等,甚至包括宏基因组、宏代谢组分析和中文帮助文档,极大了提高了流程的适用范围和易用性。
(3) USEARCH-based的扩增子分析流程。虽然已经发布了两套较完整的扩增子分析流程,但研究中存在的诸多问题却仍没有很好的解决。物理学背景的生物信息学家、独立研究员Robert Edgar在本领域开发了一系列经典的算法和软件,如高速序列比对软件USEARCH[27]、嵌合体检测软件UCHIME[28]、OTU代表性序列鉴定算法UPARSE[29]和测序数据错误过滤和去噪算法UNOISE等[30]。这些算法和软件的推出,极大的提高了扩增子数据分析的速度和准确度。在以上算法和软件的基础上,Robert逐渐将USEARCH发展成为包括近200种命令的完整扩增子分析流程,而且跨平台、体系小巧、无依赖关系和容易安装,其32位版免费,64位商业版和非赢利版分别售价1485和885美元,条件允许的实验室推荐购买,软件分析速度快且易用性强,可有效降低入门学习成本并节约宝贵时间。同时也有USEARCH类似的工具推出,如64位完全免费的VSEARCH[25],可实现USEARCH的核心功能,但下游分析功能略少。
从使用难易程度看,推荐初涉扩增子分析人员从使用USEARCH[25]或VSEARCH[25]开始,这两款软件允许用户在Windows或Mac笔记本上完成多达几百个样本的分析项目。对于有一定基础且有Linux服务器的研究者,可进一步学习QIIME 2来实现更多种类的分析方法。
统计分析和可视化部分的工作常在R语言中实现。扩增子数据分析常用R包有vegan[31]、phyloseq[32]和microbiome[33]。Vegan是群落生态包,可实现多样性、主坐标等分析,在微生物生态领域有广泛应用,甚至发展出了基于ggplot2版本的ggvegan[31]。Phyloseq[32]包的功能主要包括多样性分析、差异比较和可视化等。针对没有R使用经验的用户,phyloseq还推出了网页版工具shiny-phyloseq[34],在浏览器中即可实现扩增子数据交互式分析。Microbiome 包[33]包括多样性、核心OTU、物种组成、相关性和格式转换等80余个分析函数,提高微生物组分析的工作效率。
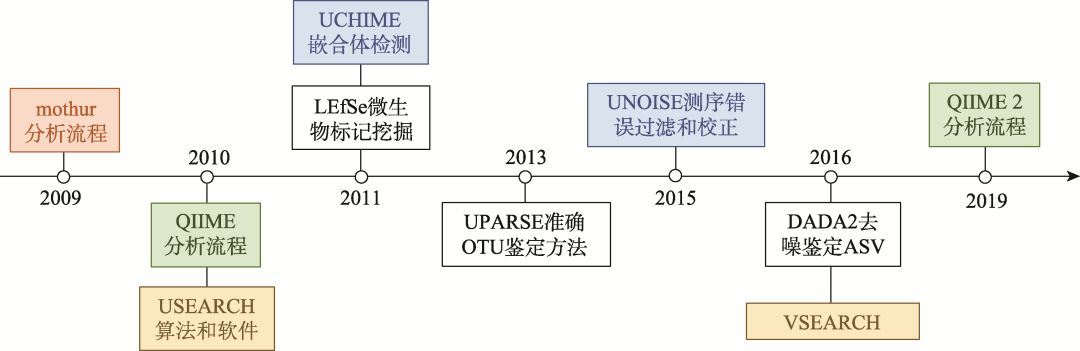
图2 近10年来微生物组领域的重要软件和算法
Fig. 2 Important softwares and algorithms of microbiome in the past decade
图中橙色为Patrick D. Schloss教授开发的分析流程mothur,绿色为Rob Knight教授主持开发的QIIME系列分析流程,蓝色显示Robert Edgar独立研究员编写的相关软件和算法。
表1 扩增子分析常用软件和数据库
Table 1 Softwares and databases for amplicon analysis
名称 | 链接 | 简介 | 参考文献 |
QIIME | http://qiime.org/ | 扩增子分析流程,功能最全、体积大、扩展性强、依赖关系多、仅限Linux或Mac系统 | [19] |
QIIME 2 | https://qiime2.org/ | 新一代扩增子分析流程,分析过程封装为压缩格式,支持分析过程全记录的可重现分析,开发并整合许多新算法处理大数据更快,可扩展性强和中文帮助文档 | [20] |
USEARCH | http://www.drive5.com/usearch/ | 比对工具,现发展为拥有200多个命令的扩增子分析流程,体积小巧、跨平台、计算速度快,但64位版收费,提供中文帮助文档(https://github.com/YongxinLiu/UsearchChineseManual) | [25] |
mothur | https://www.mothur.org/ | 最早的扩增子分析流程,体积小巧、跨平台 | [16] |
VSEARCH | https://github.com/torognes/vsearch | 扩增子分析流程,实现了USEARCH大部分的功能,喜欢USEARCH分析流程风格的替代软件,支持在QIIME 2中使用 | [25] |
Qiita | https://qiita.ucsd.edu/ | 在线扩增子分析平台,可存储数据 | [35] |
MGnify | https://www.ebi.ac.uk/metagenomics/ | 在线扩增子和宏基因组分析平台,可存储数据 | [36] |
gcMeta | https://gcmeta.wdcm.org/ | 中国科学院微生物所开发的在线扩增子和宏基因组分析平台 | [37] |
Greengenes | https://greengenes.secondgenome.com/ | 16S rRNA基因数据库,QIIME推荐数据库,但13年发表后无更新,功能注释软件PICRUSt和BugBase依赖此数据库 | [38] |
SILVA | https://www.arb-silva.de/ | rRNA基因数据库,包括真核、细菌和古菌三域的大小亚基序列,更新快、序列全,适用于物种分类和嵌合体检测 | [39] |
RDP | https://rdp.cme.msu.edu/ | 核糖体16S/28S数据库,适合物种注释,同时有在线分析流程 | [40] |
UNITE | https://unite.ut.ee/ | 真核生物ITS数据库,常用于真菌ITS扩增子测序分析中嵌合体检测和物种分类 | [41] |
vegan | https://cran.r-project.org/package=vegan | 微生物生态学领域的排序方法、多样性分析和可视化的R包,更有可视化增加的ggvegan版本https://github.com/gavinsimpson/ggvegan | [31] |
phyloseq | https://joey711.github.io/phyloseq | 扩增子分析R包,提供多样性分析、差异比较和进化树的可视化功能,同时提供网页版shiny-phyloseq | [32,34] |
microbiome | http://bioconductor.org/packages/ | 扩增子分析辅助R包,提供核心OTU/ASV计算、相关分析等函数 | [33] |
续表
名称 | 链接 | 简介 | 参考文献 |
PICRUSt | https://github.com/picrust/picrust | 基于Greengenes 16S rRNA基因预测宏基因组基因功能信息。现发布第2版实现对任意16S序列功能预测且数据库增大10倍 | [42] |
Tax4Fun | http://tax4fun.gobics.de/ | 基于SILVA 16S OTU表预测功能组成,第2版更新数据库和方法(https://sourceforge.net/projects/tax4fun2/) | [43] |
FAPROTAX | http://www.loucalab.com/archive/ | 原核分类学功能注释,获得元素循环相关文献挖掘的物种功能注释,适合于农业、环境相关研究菌种功能描述 | [44] |
BugBase | https://bugbase.cs.umn.edu/ | 物种水平微生物表型预测,如革兰氏阳/阴性、厌氧/需氧等 | [45] |
FUNGuild | http://www.stbates.org/guilds/app.php | 真菌的物种功能分类注释 | [46] |
3.2 宏基因组分析软件
近年来,鸟枪法宏基因组(shotgun metagenomic)测序随着通量提高和价格下降得到了进一步发展,随之而来的是大量相关软件的研发和发表(表2)。较扩增子测序相比,宏基因组测序不仅能获得无偏的物种组成,还得获得研究对象的功能组成,甚至能拼接出部分微生物的基因组草图。
对于人类肠道这类研究较多的领域,可选择基于参考数据库比对快速实现宏基因组物种和功能组成定量的分析方案,如MetaPhlAn2[47]、Kraken2[48]实现序列的物种分类,HUMAnN2[49]实现功能组成定量。对于缺少高质量宏基因组参考数据库的领域,则需要从头(De novo)拼接宏基因组数据,并进行基因预测。常用的宏基因组拼接软件有MEGAHIT[70]和metaSPAdes[50]等,基因注释软件如Prokka[51]和GeneMarkS-2[52]等(表2)。对于多样品或多批次的宏基因组数据进行合并分析,通常还要采用CD-HIT[53]构建非冗余基因集(non-redundancy gene catalog),实现将所有样本基于统一的参考序列进行定量和比较。获得的基因集比对至多种蛋白功能注释数据库,提供更多角度观察数据的生物学意义,如常用的数据库有碳水化合物基因数据库CAZy[54]、抗生素抗性基因综合数据库CARD[55]和毒力因子数据库VFDB[56]等。
表2 宏基因组分析常用软件和数据库
Table 2 Metagenome analysis softwares and databases
名称 | 链接 | 简介 | 参考文献 |
MultiQC | https://multiqc.info/ | 多样本质控和分析结果汇总 | [66] |
Trimmomatic | http://www.usadellab.org/cms/index.php? | Java编写的质量控制软件,实现快速去除低质量、接头和引物序列。被质控流程KneadData流程整合为默认质控软件。 | [67] |
Bowtie 2 | http://bowtie-bio.sourceforge.net/bowtie2 | 序列比对工具,短读长序列快速比对至参考序列,结果为SAM/BAM格式 | [68] |
MetaPhlAn2 | https://bitbucket.org/biobakery/ | 物种组成定量流程,包括人工整理的上万物种中的上百万个标记基因数据库,结果可直接用于LEfSe分析 | [47] |
HUMAnN2 | https://bitbucket.org/biobakery/humann2 | 功能组成定量流程,默认基于UniRef数据库注释序列,获得基因家族、通路丰度和覆盖度的功能组成表 | [49] |
UniRef | https://www.uniprot.org/uniref/ | 非冗余蛋白序列数据库,用于宏基因组分析中序列或基因的功能注释 | [69] |
Kraken 2 | https://ccb.jhu.edu/software/kraken2/ | 物种分类软件,基于K-mer方式匹配NCBI 非冗余数据库实现超高速物种注释,内存要求高 | [48] |
MEGAHIT | https://github.com/voutcn/megahit | 宏基因组拼接软件,内存消耗低,计算速度快、嵌合体率较低、N50偏低 | [70] |
metaSPAdes | http://cab.spbu.ru/software/spades/ | 宏基因组拼接软件,内存消耗大,计算时间长,但有更长的N50,也存在拼接错误和嵌合体比例升高的风险 | [50] |
MetaQUAST | http://quast.sourceforge.net/metaquast | 拼接结果评估,输出拼接指标和可视化图形的PDF和交互式网页版报告 | [71] |
Prokka | http://www.vicbioinformatics.com/ | 原核基因组注释流程,主要用于基因组、宏基因组中的编码基因预测,生成提交NCBI所需要的注释文件 | [51] |
GeneMarkS-2 | http://exon.gatech.edu/GeneMark/ | 基因组注释网页工具,用户无需服务器和安装软件,浏览器中实现宏基因组中基因预测 | [52] |
CD-HIT | http://weizhongli-lab.org/cd-hit/ | 序列去冗余,实现核酸、蛋白构建非冗余基因集 | [53] |
Salmon | https://combine-lab.github.io/salmon/ | 非比对基因定量,基于K-mer方式超快速实现序列分配,无中间文件生成,直接获得计数型结果 | [72] |
DIAMOND | https://github.com/bbuchfink/diamond | 比BLAST更快的蛋白比对工具 | [73] |
eggNOG | http://eggnogdb.embl.de/app/emapper#/ | 同源组蛋白数据库 | [74] |
GhostKOALA | https://www.kegg.jp/ghostkoala/ | 在线KEGG注释工具,可为基因序列分配KO编号 | [75] |
CAZy | http://www.cazy.org/ | 蛋白功能注释:碳水化合物基因数据库 | [54] |
CARD | https://card.mcmaster.ca | 蛋白功能注释:抗生素抗性基因综合数据库 | [55] |
Resfams | http://www.dantaslab.org/resfams | 蛋白功能注释:抗生素抗性基因数据库 | [76] |
VFDB | http://www.mgc.ac.cn/VFs/ | 蛋白功能注释:毒力因子数据库 | [56] |
MetaBAT 2 | https://bitbucket.org/berkeleylab/metabat/ | 主流分箱工具 | [57] |
MaxBin 2 | https://sourceforge.net/projects/maxbin2/ | 主流分箱工具 | [58] |
CONCOCT | https://github.com/BinPro/CONCOCT | 主流分箱工具 | [59] |
metaWRAP | https://github.com/bxlab/metaWRAP | 分箱流程,依赖140余款工具,可实现conda快速安装,默认对3种主流分箱结果提纯,提供多种可视化方案 | [60] |
DAS_Tool | https://github.com/cmks/DAS_Tool | 分箱流程,对5种主流分箱工具结果提纯 | [61] |
续表
名称 | 链接 | 简介 | 参考文献 |
Athena | https://github.com/elimoss/metagenomics_workflows/ | 基于10×建库宏基因组测序的组装软件 | [63] |
OPERA-MS | https://github.com/CSB5/OPERA-MS | 基于Illumina、Nanopore和PacBio的二、三测序数据混合组装软件 | [64] |
MAGpy | https://github.com/WatsonLab/MAGpy | 分箱结果下游比较基因组分析流程 | [65] |
OrthoFinder | https://github.com/davidemms/ | 同源基因鉴定,基于多个细菌基因组中的蛋白组鉴定单拷贝同源基因和构建多基因进化树 | [77] |
Microbiome helper | https://github.com/LangilleLab/ | 微生物组分析中常用格式转换工具集,方便分析和流程搭建 | [78] |
宏基因组测序除了可以揭示研究对象的物种和功能组成外,还可能通过分箱(binning)方法组装出单菌基因组。近年来分箱软件快速发展,使获得不可培养微生物的基因组成为可能。目前常用的分箱工具有MetaBAT 2[57]、MaxBin 2[58]和CONCOCT[59]等,但结果差别较大。去年发表了两款分箱提纯工具metaWRAP[60]和DAS_Tool[61]解决了分箱工具选择难、结果差异大的问题,他们通常整合3~5款分箱工具的结果,进一步筛选和综合利用,获得更高质量的单菌基因组,同时提供分箱的定量、注释等一系列常用分析功能。值得注意的是,分箱获得的单菌基因组存在着不完整和高污染等问题,因此想要提高宏基因组中单菌组装的完整性,从实验手段进行改进并采用配套专用分析方法是未来的发展方向,如采用流式细胞术单细胞分选[62]、10×建库[63]、二三代混合测序[64,65]等新方法在宏基因组拼接和分箱中取得了较好的效果。宏基因组分析中常用的软件和数据库简介详见表2。
3.3 统计和可视化工具
扩增子和宏基因组分析获得的物种和功能组成表统称为特征表,是第二代测序数据分析结果中的通用格式,在下游分析中可以通过选择多种R包、图形化界面、命令行或网页版工具进行数据的转换和呈现。Bioconductor网站提供了上千种生物学数据分析R包,例如计数型数据可选基于负二项分布模型的差异统计R包edgeR[79]或DESeq2[80],组成型数据差异分析可选limma包[81],结合已知影响因素数据校正的差异比较可选支持广义线性混合效应模型的lme4包[82]。STAMP是为微生物组数据开发的跨平台、图形界面统计分析工具[83],可以实现主成分分析、多种统计方法进行两组或多组差异比较,结果可选散点图、箱线图、柱状图、热图和扩展柱状图等展示方法。LEfSe可以实现基于线性判别分析寻找特征向量的命令行工具[84],结果可选柱状图和基于GraPhlAn绘制的进化分枝图(Cladogram)等展示方式[85],没有Linux服务器或不熟悉命令行工作的研究者还可以选择网页版LEfSe开展分析。此外,还有一些专门收集整理微生物组工具并提供在线分析和可视化的平台,让用户在浏览器中即可完成分析工作,例如MicrobiomeAnalyst[86]可实现基于特征表和元数据进行数据筛选、标准化、多样性分析、差异比较和机器学习等多种分析和可视化方案。
3.4 网络分析
网络分析是一门基于图论的学科,因其独特的视角和直观的可视式结果在微生物组数据分析中也有广泛的应用。2018年,FEMS Microbiology Review发表综述文章系统介绍了目前主流网络分析方法的优缺点、适用范围和选择依据[87];Nature Reviews Microbiology发表综述文章介绍了网络图在群落结构研究中的作用和意义[88];此外,陈亮2017年在宏基因组公众号发布的《Co-occurrence网络图在R中的实现》对相关基础概念和具体的实现方法进行了介绍,也可供学习参考。常用的分析方法有网页工具MENAP[89],本地相似分析LSA[90]、专为微生物组稀疏型数据开发的相关性算法SPARCC[91]、作为Cytoscape[92]插件使用的CoNet[93]、R语言中的WGCNA[94]和SpiecEasi[95]包等。具体的操作也比较容易实现,例如在R语言环境中使用WGCNA[94]包计算网络相关性质,采用igraph[96]包实现网络的可视化。对于网络的进一步分析、可视化细节调整,可将网络数据导入Cytoscape[92]或Gephi[97]中调整细节。目前该分析已在pH与微生物群落组装[98]、妊娠糖尿病与健康孕妇微生物组结构、洗牙后口腔微生物群落结构恢复等研究中得到应用[99,100]。
3.5 进化分析
微生物组数据非常适合开展进化分析,因为单物种的研究需要搜集和整理大量相关研究中的同源基因,而微生物组研究中的扩增子测序可获得的序列就是成千上万的同源基因,方便开展物种系统发育关系研究。进化分析主要分为多序列对齐、进化树构建和进化树美化等3个基本过程。由于微生物组中序列种类多且复杂度高,需要选择计算速度快的工具。多序列对齐可采用MAFFT[101]或MUSCLE[102];进化树构建可选FastTree[103]或IQ-TREE[104,105];最后采用Evolview[106]或iTOL[107]在线进行进化树的可视化和美化。推荐将序列对应的物种和丰度信息表使用R脚本table2itol (https://github.com/mgoeker/ table2itol)格式化为iTOL的输入文件。此外,R语言中的ggtree包也可以实现进化树的注释和美化[108]。展示物种注释层级结构的进化分枝图(Cladogram),推荐使用GraPhlAn进行可视化[85]。宏基因组测序是鸟枪法随机片段测序,进化分析需要采用OrthoFinder[77]基于分箱结果鉴定单拷贝同源基因,并构建多基因进化树。
3.6 机器学习
机器学习是当前计算机算法研究中最热门的领域,专门研究计算机如何模拟或实现人类的学习行为,以获取新的知识或技能,重新组织已有的知识结构使之不断改善自身的性能[109]。目前在微生物组领域常用的机器学习方法有随机森林(Random Forest)、支持向量机(support vector machine, SVM)和Adaboost等。其中随机森林分类(Classification)在饮食习惯分型[110]、疾病诊断[111]、植物亚种预测[9]等领域有较多应用;随机森林回归(Regression)在婴儿营养健康[2]、法医学[112]、时间序列预测[113]等领域有广泛的应用。开展随机森林分析可在R语言中通过使用randomForest包实现[114]。深度学习是机器学习领域新发展的方法,最近预印本服务器BioRxiv在线发表了基于肠道菌群数据的深度学习可准确预测人类真实年龄[115],此项研究还被Science杂志新闻报导。
3.7 其他分析工具
许多其他领域的分析方法在微生物组中也得到了推广和应用。全基因组关联分析(genome-wide association study, GWAS)[116]在鉴定人类疾病相关基因中发挥了巨大作用,目前也应用于微生物组领域来大规模探索人类与微生物组间的调控规律[117,118]、植物微生物组与产量[119]等。环境因子关联分析也有较多的分析方法在微生物生态学中得到广泛应用,如揭示温度[120]、pH[121]和盐分[122]等在不同环境中是微生物群落结构的决定因素。更多关于微生物组下游分析工具的介绍,详见表3。
4 分析代码重用
很多文章中的分析和可视化结果并非基于发表软件,而且作者自编程实现的分析。如果想参考文章中的分析方法和图表,根据方法描述自行组合工具或编写代码是非常有挑战的工作。目前很多文章发表时提供了分析代码,链接位于文章“代码可用(Code Available)”栏目,代码保存于Github等代码备份网站。基于文章作者分享的代码和测试数据,
更容易重复文章中发表的分析方法,在理解的基础上替换为自己的数据开展分析,甚至可在源代码基础上修改分析方案,获得更合理的结果。分析代码的重现性在研究中可极大地提高工作效率,节省研究者大量开发分析代码的时间。表4列举了一些提供可重复分析代码的实验室,供研究者参考。
表3 微生物组下游通用分析工具
Table 3 Downstream softwares for microbiome analysis
名称 | 链接 | 简介 | 参考文献 |
edgeR | http://bioconductor.org/packages/edgeR/ | 数字基因表达数据的经验分析R包,常用于基于计数型数据和负二项分布模型进行差异统计 | [79] |
DESeq2 | http://bioconductor.org/packages/DESeq2/ | 基于负二项分布的差异基因表达分析R包,与edgeR包类似 | [80] |
limma | http://bioconductor.org/packages/limma/ | 基于线性模型分析芯片数据R包,可用于微生物组数据差异比较 | [81] |
lme4 | https://github.com/lme4/lme4/ | 拟合线性和广义线性混合效应模型,可结合已知影响因素数据校正的差异比较 | [82] |
STAMP | http://kiwi.cs.dal.ca/Software/STAMP | 图型界面的微生物组统计与可视化软件,跨平台,Windows中安装方便,但不支持中文,Linux/Mac中安装困难 | [83] |
LEfSe | https://bitbucket.org/biobakery/biobakery/wiki/lefse | 微生物组生物标记挖掘工具,支持Linux命令行、网页界面、多组比对,结果可视化为柱状图和进化分枝图 | [84] |
GraPhlAn | https://bitbucket.org/nsegata/graphlan | 进化分枝图可视化工具 | [85] |
MicrobiomeAnalyst | http://www.microbiomeanalyst. | 在线微生物组特征表分析平台,支持几十种常用分析和可视化,可导出网页版分析报告 | [86] |
igraph | https://igraph.org/r/ | 网络图可视化平台,可在R语言中可实现网络图可视化、布局和细节调整 | [96] |
Cytoscape | https://cytoscape.org/ | 网络分析和可视化图型界面分析平台,功能强大,跨平台,扩展插件丰富 | [92] |
Gephi | https://gephi.org/ | 网络分析和可视化软件,样式比较美观 | [97] |
MAFFT 7 | https://mafft.cbrc.jp/alignment/ | 多序列对齐软件,序列对齐速度快 | [101] |
MUSCLE | https://www.drive5.com/muscle/ | 多序列对齐软件,序列对齐速度快 | [102] |
IQ-TREE | http://www.iqtree.org/ | 进化树构建,在运行速度上有较明显的优势,跨平台,速度快,提供在线版 | [104,105] |
iTOL | https://itol.embl.de/ | 进化树可视化、编辑和美化工具,功能全面,支持结果生成分享链接 | [107] |
randomForest | https://cran.r-project.org/web/ | 实现随机森林分类和回归分析的R包 | [114] |
表4 部分提供统计分析代码的实验室
Table 4 Labs that provide statistical analysis codes
研究单位 | 课题组 | 链接 | 参考文献 |
美国密歇根大学 | Patrick D. Schloss | http://www.schlosslab.org | [123] |
美国斯坦福大学 | Susan Holmes | http://statweb.stanford.edu/~susan | [124] |
德国马普植物育种研究所 | Paul Schulze-Lefert | https://github.com/garridoo | [125] |
美国北卡罗来纳大学教堂山分校 | Jeffery L. Dangl | https://github.com/surh/pbi | [126,127] |
EMBL-EBI | Robert D. Finn | https://github.com/Finn-Lab | [128] |
续表
研究单位 | 课题组 | 链接 | 参考文献 |
比利时鲁汶大学 | Jeroen Raes | https://github.com/raeslab | [129] |
美国贝勒医学院 | Christopher J. Stewart | https://github.com/StewartLab | [130] |
美国俄勒冈大学 | James F. Meadow | https://github.com/jfmeadow | [131] |
中国科学院遗传与发育生物学研究所 | Yang Bai | https://github.com/microbiota | [132,133] |
5 结语与展望
近10年来,第二代测序技术通量的提高和价格的下降,极大地推动了微生物组领域的发展,使得研究者拓宽了微生物组研究对象的深度和广度,揭示了极端环境、植物、动物、人类肠道、海洋、土壤等领域的微生物组成和功能[6]。目前宏基因组研究主要以短读长的Illumina Seq/Nova系列或华大基因的BGI Seq系列平台产出数据为主,虽然获得数据通量大,但数据拼接质量仍有较大提升空间。近年来,Pacific BioSciences (PacBio)和Oxford Nanopore Technologies (ONT)等三代测序技术快速发展,虽然受到测序错误率高和配套软件缺乏的困扰,但在读长、测序速度等方面的优势正在逐渐突显。Charalampou等[134]应用ONT技术对患者呼吸道细菌宏基因组进行测序,实现了6 h内快速诊断致病菌。
目前微生物组研究中应用最广泛的是扩增子测序技术,该技术可以快速地揭示群落的微生物组成,而且具有操作简单、成本低、有效避免宿主污染、方便开展大规模研究等优势。但扩增子的研究范围仅限引物可扩增部分DNA的物种组成,而且受扩增基因拷贝数和多态性的影响,如果想进一步了解微生物组的全貌和功能基因,宏基因组是更有效的研究方法。宏基因组不仅可以无偏的获得研究对象中细菌、真菌、古菌、病毒和原生动物等一切以DNA为遗传物种的物种序列信息、确定其物种和功能组成,更有潜力获得未培养物种的功能基因,甚至是基因组草图。目前虽然已经有一些宏基因组分箱、分箱提纯的工具,但仍处于发展的初级阶段,还有很多有待改进的方向,如计算不同长度K-mer频率、比对参考数据库去除已知物种降低复杂度和/或结合三代长读长的测序数据等[64,135]。
提高微生物组数据分析的效率,高质量的参考数据库是基础,而这一领域的发展依赖于大规模培养组学的应用和更多高质量参考基因组的公布。同时,对发表数据的分类整理、提高可用性以及进一步挖掘也十分必要。例如,R包curatedMetagenomicData整理了46个研究中的8184个宏基因组样本,对超100 TB的原始数据采取了严格质控进而获得了相关物种和功能组成表,方便同领域研究者对数据进一步挖掘和查询[136];ML Repo数据库整理来自15篇文章中的33个人类微生物组IBD、糖尿病、肥胖和癌症等分类和年龄回归数据集,研究者可按类浏览下载这些数据,用于进一步挖掘和方法评估[137];意大利特伦托大学Nicola Segata团队利用来自不同地理位置、生活方式和年龄人群的9428个宏基因组,突破性地重建了15万个人体微生物基因组草图[138]。以上对发表数据整理和再利用的例子,为今后开发更多基于发表数据的数据库和分析工具提供了借鉴和参考。
参考文献(References):共138篇,略,详见原文。请点击阅读原文下载全文。
(责任编委: 赵方庆)
猜你喜欢
10000+:菌群分析 宝宝与猫狗 梅毒狂想曲 提DNA发Nature Cell专刊 肠道指挥大脑
文献阅读 热心肠 SemanticScholar Geenmedical
16S功能预测 PICRUSt FAPROTAX Bugbase Tax4Fun
生物科普: 肠道细菌 人体上的生命 生命大跃进 细胞暗战 人体奥秘
写在后面
为鼓励读者交流、快速解决科研困难,我们建立了“宏基因组”专业讨论群,目前己有国内外5000+ 一线科研人员加入。参与讨论,获得专业解答,欢迎分享此文至朋友圈,并扫码加主编好友带你入群,务必备注“姓名-单位-研究方向-职称/年级”。PI请明示身份,另有海内外微生物相关PI群供大佬合作交流。技术问题寻求帮助,首先阅读《如何优雅的提问》学习解决问题思路,仍未解决群内讨论,问题不私聊,帮助同行。

学习16S扩增子、宏基因组科研思路和分析实战,关注“宏基因组”
 点击阅读原文,跳转文章主页下载全文
点击阅读原文,跳转文章主页下载全文

















 5741
5741

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









