






写在前面
之前介绍过使用EggNOG-mapper对非模式物种快速完成KEGG注释的推文https://mp.weixin.qq.com/s/kIf6C2u3FID3ZeLtsB4eZQ,不过毕竟EggNOG-mapper是第三方软件,而且注释结果仍然存在一个Gene ID对应多个KO Number的情况,虽然对富集结果影响似乎不会太大,但是影响多少肯定是会有。
这里介绍KEGG官方提供的注释工具KofamKOALA,同样能够对任意物种进行KEGG注释,注释结果中Gene ID对应唯一一个KO Number,避免了大多数第三方注释工具一个ID对应多个KO Number的情况,使富集结果更加可靠!
本教程含「在线版」和「本地版」,满足各类注释场景。
KofamKOALA官网https://www.genome.jp/tools/kofamkoala/
一、网页版KofamKOALA
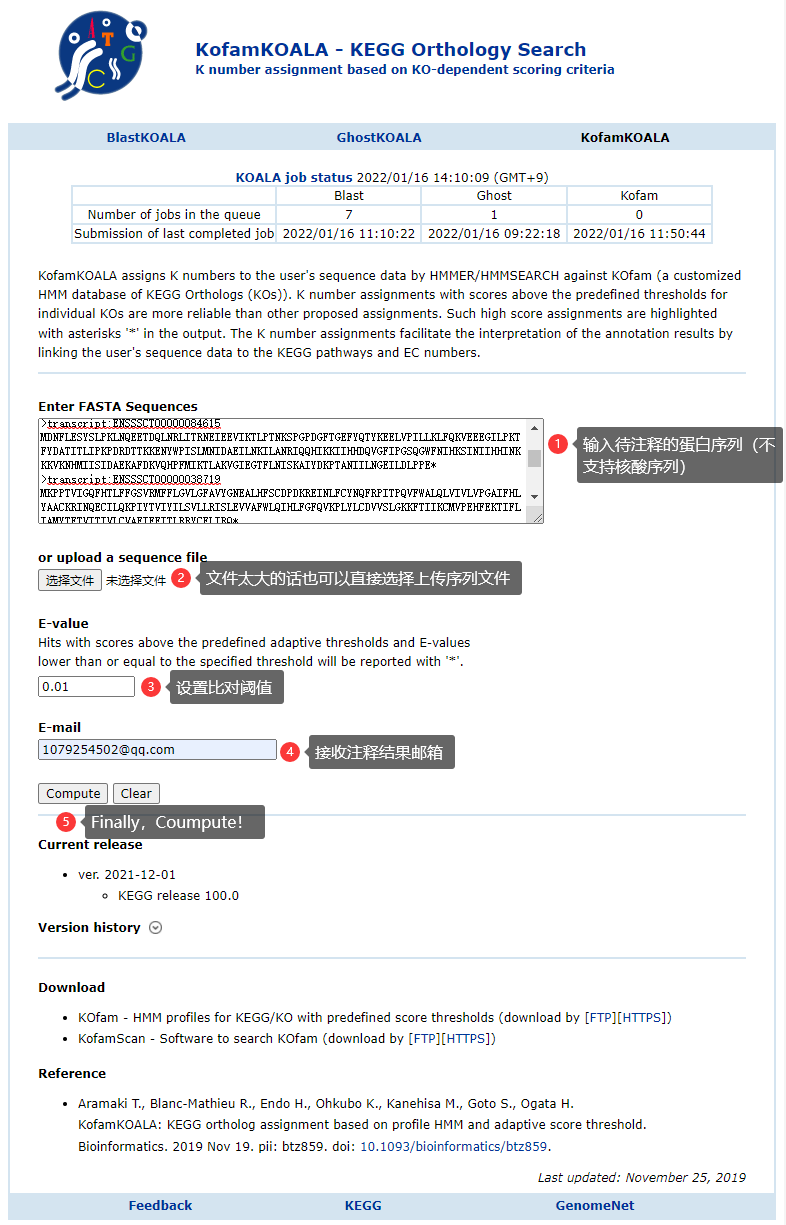
点击提交任务之后需要在邮件中二次确认,提交任务
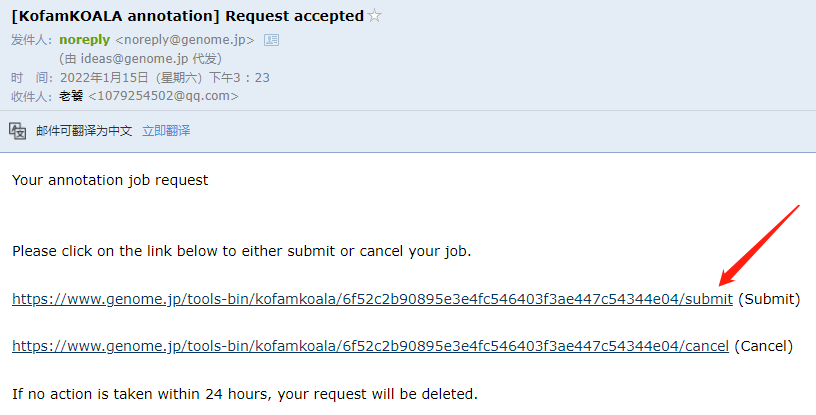
注释完成之后,点击邮件中的链接

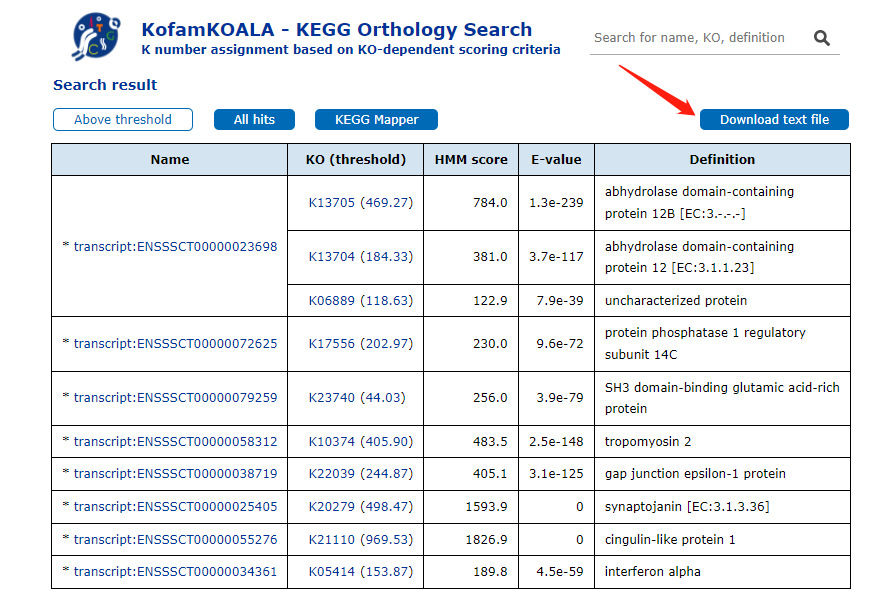
在结果页面中,可以直接下载注释结果,然后接着用TBtools进行富集分析
同时也提供了许多对注释结果的探索功能,比如查看注释上的KO Number详细信息,比对情况等。
二、本地化KofamKOALA
如果需要注释的序列太多,文件太大,使用网页版工具确实可能会太慢。这个时候当然是选择将其本地化!在本地根据服务器情况提高线程加速注释。
下载 KOfam 和 KofamScan
下载并解压KOfam
wget https://www.genome.jp/ftp/db/kofam/ko_list.gz
wget https://www.genome.jp/ftp/db/kofam/profiles.tar.gz
#下载完成之后解压
gunzip ko_list.gz
tar -xzvf profiles.tar.gz下载并解压kofam_scan
wget https://www.genome.jp/ftp/tools/kofam_scan/kofam_scan-1.3.0.tar.gz
tar -xzvf kofam_scan-1.3.0.tar.gz将kofam_scan加入环境变量
echo export PATH=/home/jiawei_li/tools/kofamscan/kofam_scan-1.3.0:\$PATH >> ~/.bashrc
source ~/.bashrc安装依赖,KofamScan需要Ruby,HMMER和GNU Parallel
#版本需求
- Ruby >= 2.4
- HMMER >= 3.1
- GNU Parallelconda安装Ruby,自己编译也行 但是我比较懒折腾。。。
conda install -c conda-forge ruby如果喜欢手动装Ruby,也可以试试,但也许会报错,需要进行一系列操作去安装
wget https://cache.ruby-lang.org/pub/ruby/3.1/ruby-3.1.0.tar.gz
tar -xzvf ruby-3.1.0.tar.gz
cd cd ruby-3.1.0
./configure
make
make install安装hmmer
conda install -c bioconda hmmer安装GNU Parallel
conda install -c conda-forge parallel修改配置文件,指定依赖软件以及KOfam的路径
首先获取相关软件的路径(注意:配置文件中不需要指定Ruby的路径,但是需要确保Ruby加入了环境变量,能够直接调用)
which parallel hmmscan
#/tools/parallel
#~/miniconda3/bin/hmmscan使用官方模板,修改配置文件
cd kofam_scan-1.3.0
cp config-template.yml config.yml注意,由于hmmscan和parallel都已经加入环境变量,可以直接调用,因此在配置文件中无需配置这两个软件的路径
# Path to your KO-HMM database
# A database can be a .hmm file, a .hal file or a directory in which
# .hmm files are. Omit the extension if it is .hal or .hmm file
profile: /home/jiawei_li/tools/kofamscan/profiles
# Path to the KO list file
ko_list: /home/jiawei_li/tools/kofamscan/ko_list
# Path to an executable file of hmmsearch
# You do not have to set this if it is in your $PATH
#hmmsearch: /home/jiawei_li/miniconda3/bin/hmmscan
# Path to an executable file of GNU parallel
# You do not have to set this if it is in your $PATH
#parallel: /tools/parallel
# Number of hmmsearch processes to be run parallelly
cpu: 8对蛋白序列进行注释(注意:注释序列必须为蛋白序列)
exec_annotation -o test.querry2KO --cpu 8 --format mapper -E 1e-5 test.pep.fa注意: --format参数在help中给出了四种,但主要有两种格式。
#help文档中的解释
-f, --format <format> Format of the output [detail]
detail: Detail for each hits (including hits below threshold)
detail-tsv: Tab separeted values for detail format
mapper: KEGG Mapper compatible format
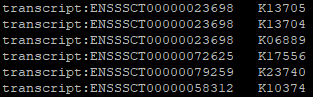
mapper-one-line: Similar to mapper, but all hit KOs are listed in one line--format mapper对每一个Gene ID只保留最佳的KO Number,结果文件只含有Gene ID和KO Number的映射信息。
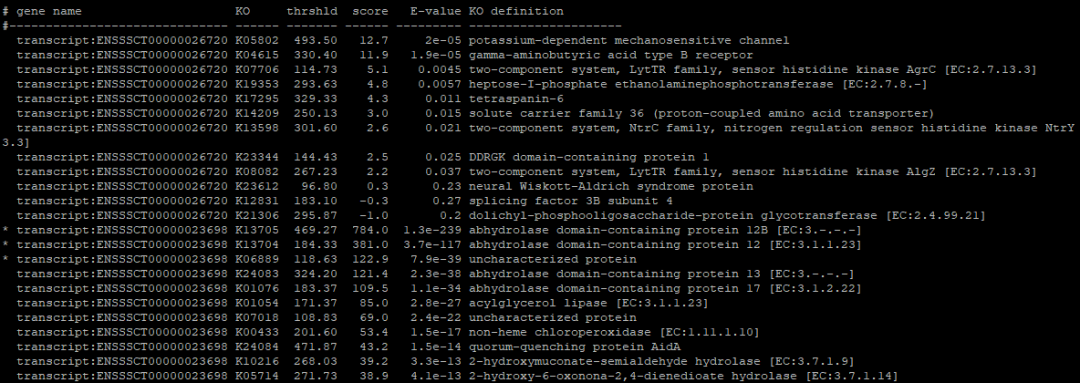
--format detail保留所有信息,包括Gene ID对应上的每一个KO Number,比对分数,E-value以及KO Number的详细信息等。
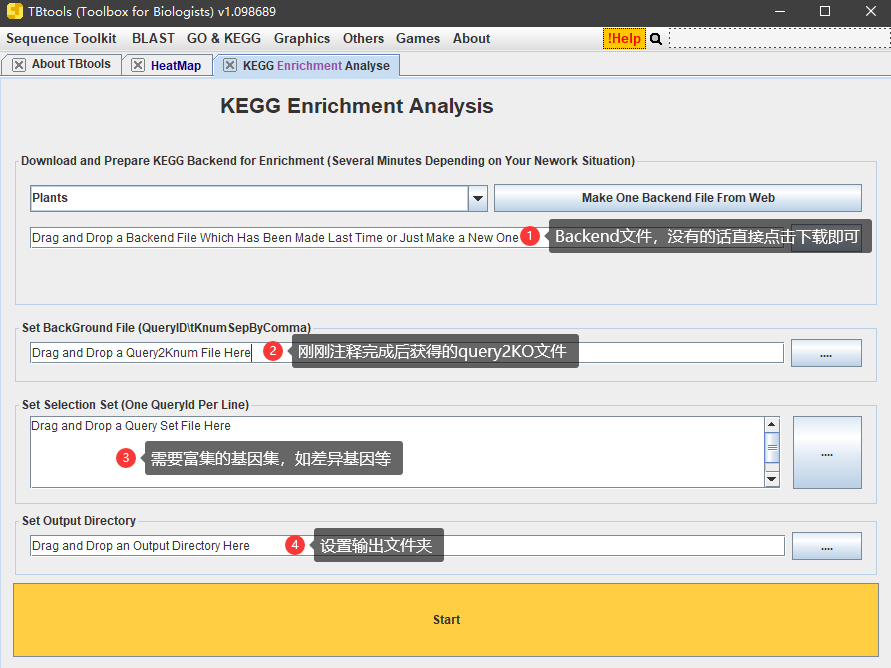
最后,拿到query2KO文件之后,就可以用其当背景文件,使用TBtools对基因集做富集分析啦
猜你喜欢
iMeta简介 高引文章 高颜值绘图imageGP 网络分析iNAP
iMeta网页工具 代谢组MetOrigin 美吉云乳酸化预测DeepKla
iMeta综述 肠菌菌群 植物菌群 口腔菌群 蛋白质结构预测
10000+:菌群分析 宝宝与猫狗 梅毒狂想曲 提DNA发Nature
一文读懂:宏基因组 寄生虫益处 进化树 必备技能:提问 搜索 Endnote
16S功能预测 PICRUSt FAPROTAX Bugbase Tax4Fun
生物科普: 肠道细菌 人体上的生命 生命大跃进 细胞暗战 人体奥秘
写在后面
为鼓励读者交流快速解决科研困难,我们建立了“宏基因组”讨论群,己有国内外6000+ 科研人员加入。请添加主编微信meta-genomics带你入群,务必备注“姓名-单位-研究方向-职称/年级”。高级职称请注明身份,另有海内外微生物PI群供大佬合作交流。技术问题寻求帮助,首先阅读《如何优雅的提问》学习解决问题思路,仍未解决群内讨论,问题不私聊,帮助同行。
点击阅读原文,跳转最新文章目录阅读

















 4344
4344

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









