









CUT&Tag是研究蛋白和DNA互作的新兴实验方法,该方法是通过蛋白特异性抗体引导Protein A-Tn5酶在目标蛋白结合的DNA位置进行切割并且在序列两端加上测序接头,经过PCR扩增后形成可以用于高通量测序的文库,随后对文库进行测序分析从而解析与目标蛋白结合的DNA片段。
利用CUT&Tag,我们可以得到目标转录因子/辅因子/组蛋白结合位点信息,确定靶基因信息;对相关基因组功能元件(诸如启动子、增强子)进行注释分析;所需样本起始量低,适合珍惜样本(如胚胎细胞、数量有限的原代细胞、流式分选后的细胞等)。
单一组学分析可能无法完全揭示生物系统的复杂性,多种组学数据的整合可以通过构建生物分子网络和路径,揭示生物系统的整体性和系统性特征。CUT&Tag作为 2019年报道的蛋白和DNA互作的实验方法,发表过的研究已广泛应用多组学,其中最为普遍是“CUT&Tag+RNA-seq”组合。
CUT&Tag和RNA-seq关联分析也比较套路,一般有以下几种。
1. CUT&Tag关联基因和RNA-seq的差异表达基因进行overlap。
ChIP-seq数据差异分析拿到关联基因的基因集,与RNA-seq数据的差异表达基因的数据集取交集,这个结果通常用于筛选关键目的基因。一般是通过venn图进行可视化展示。网上有很多在线工具可以绘制venn图,当然爱基百客的云平台也提供venn图绘制的小工具。
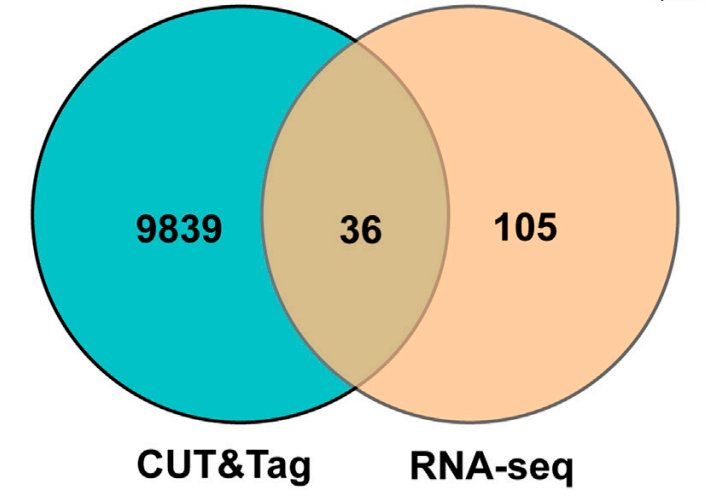
图:CUT&Tag和RNA-seq数据关联分析匹配基因[1]
爱基百客云平台小工具
2. 对overlap过的基因进行功能分析。
功能分析可以分为通用的GO/KEGG分析,也可以根据已有的文献报道进行挖掘。
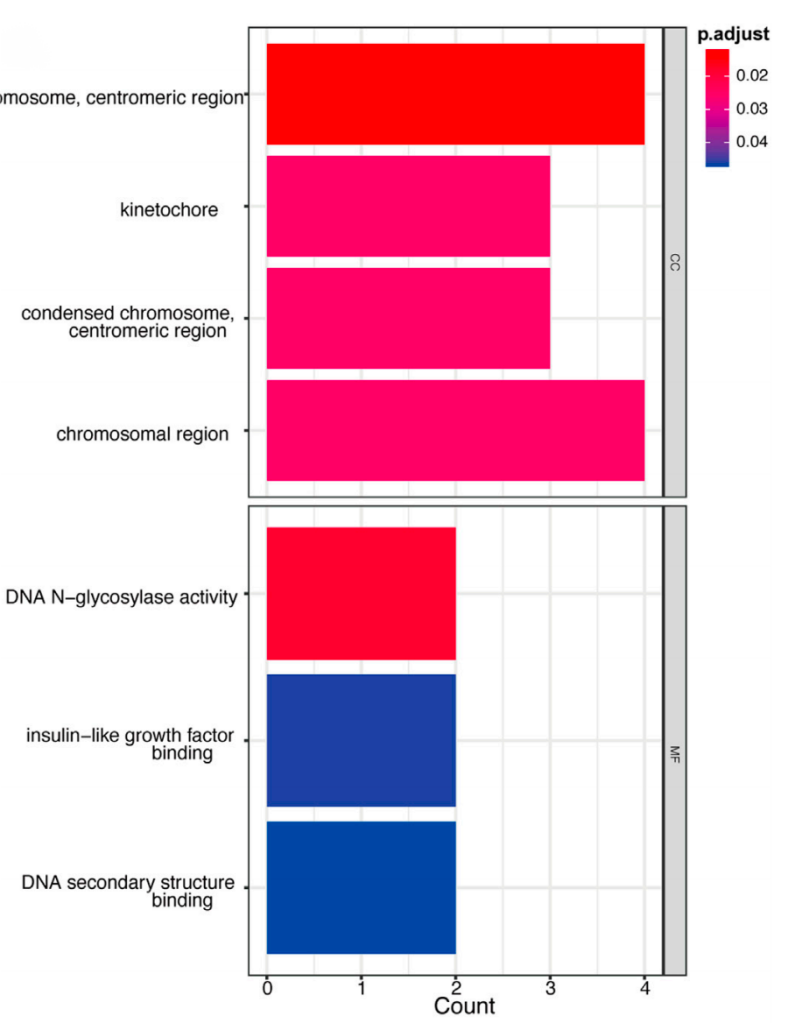
overlap后的匹配的差异基因的GO分析[1]
3. 基因组浏览器可视化
确定目标基因后,可以对相关基因的mRNA水平和Peak进行联合可视化展示,结果类似下图。这一类结果通常是利用基因组浏览器(如IGV、UCSC Genome Brower等)展示特定区域的CUT&Tag和RNA-seq数据,直观观察转录因子结合位点、组蛋白修饰的分布和水平以及相应基因的表达水平。
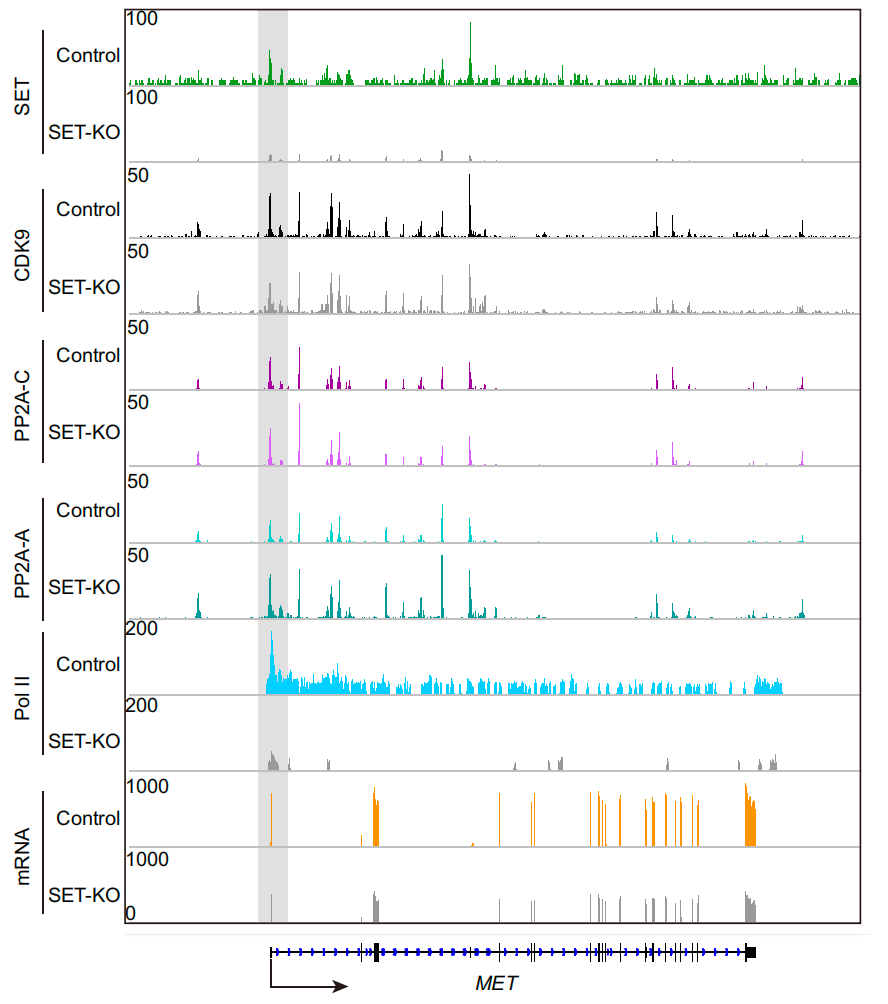
图:在MET基因座位上,叠加显示了CUT&Tag、ChIP-seq和RNA-seq的轨迹,展示了SET、CDK9、PP2A-A、PP2A-C和Pol II在染色质上的占据情况,以及在对照组和SET-KO细胞中的mRNA表达情况[2]
4. 基因调控网络图
利用软件工具如Cytoscape等,根据CUT&Tag和RNA-seq数据构建的基因表达调控网络,直观地展示转录因子、组蛋白修饰与目标基因之间的互相作用。
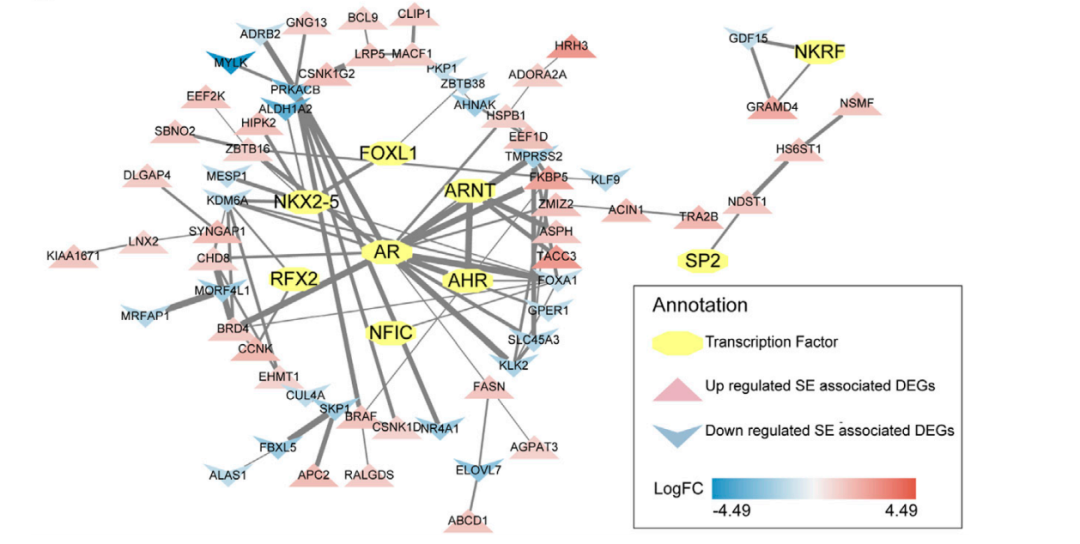
图:预测的转录因子及其与超级增强子SE相关的差异表达基因的相互作用。[3]
5. 相关性分析
将基因表达水平的差异倍数和相关peak信号强度差异进行相关性分析。以下图为例,结果展示了基因表达变化与H3K36me3信号之间的相关性,挖掘其中调控的关系。
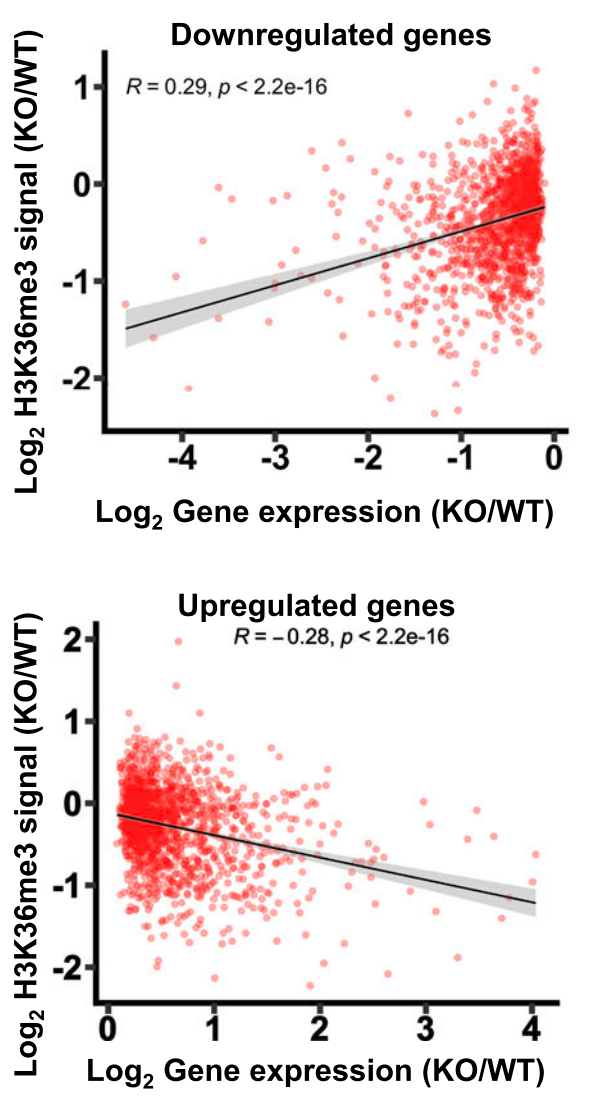
图:基因表达变化与H3K36me3信号之间的相关性[4]
爱基百客专注于表观组学10年,提供领先的表观组学服务。在CUT&Tag上拥有丰富的经验,提供从方案设计、样本处理、建库测序、数据分析和验证一站式服务;另外,有云平台,多组学联合分析助力深度挖掘数据。目前,CUT&Tag正在做春季大促,有相关需求的老师欢迎咨询驻地销售或联系助理小爱。

CUT&Tag产品介绍
CUT&Tag是研究蛋白和DNA互作的新兴实验方法,该方法是通过蛋白特异性抗体引导Protein A-Tn5酶在目标蛋白结合的DNA位置进行切割并且在序列两端加上测序接头,经过PCR扩增后形成可以用于高通量测序的文库,随后对文库进行测序分析从而解析与目标蛋白结合的DNA片段。
产品优势:
1. 无需甲醛交联,样本要求量低,背景干净,可重复性好;
2. 提供从方案设计,建库测序,到数据分析和验证一站式服务;
3. 云平台,多组学联合分析深度挖掘数据;
4. 项目经验丰富:

实测数据:
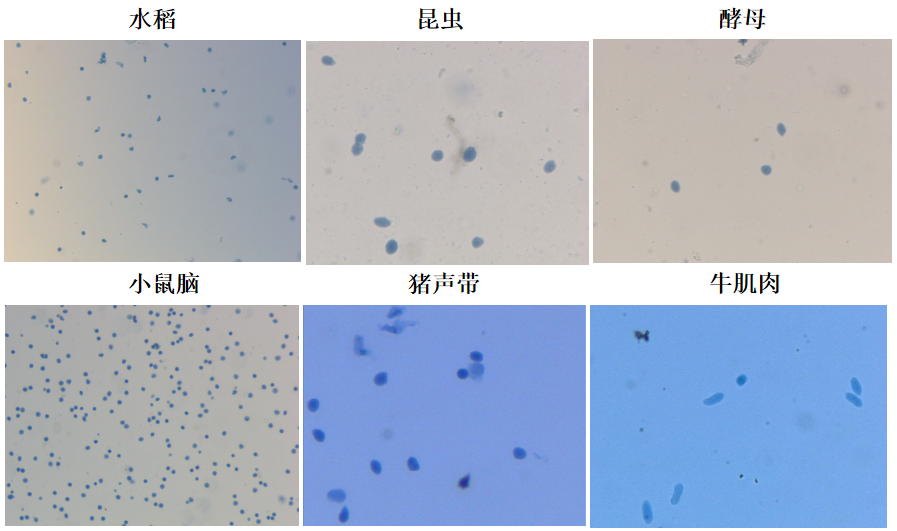
1. 抽核结果
提供“单细胞测序”标准的抽核解决方案。
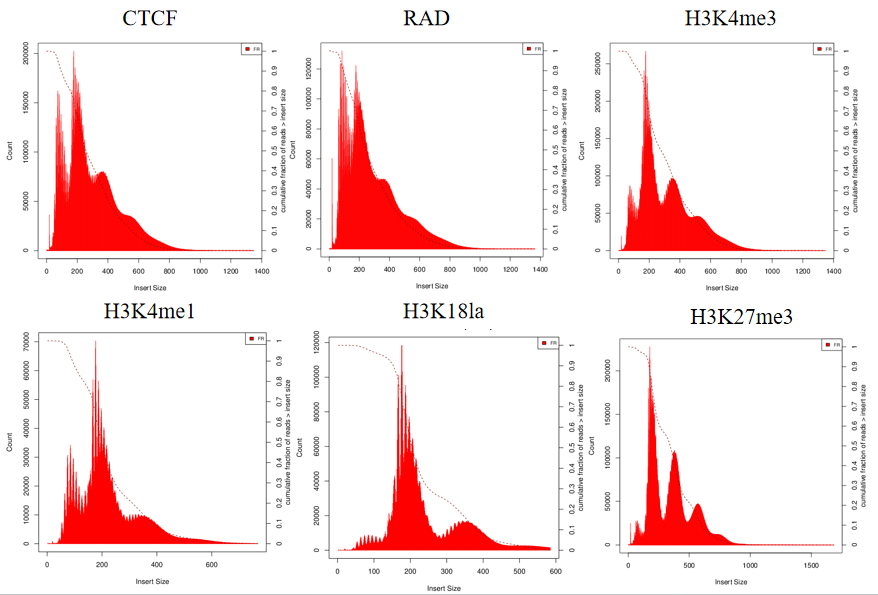
2. CUT&Tag酶切片段分布
转录因子/组蛋白修饰的酶切片段分布呈现周期性。
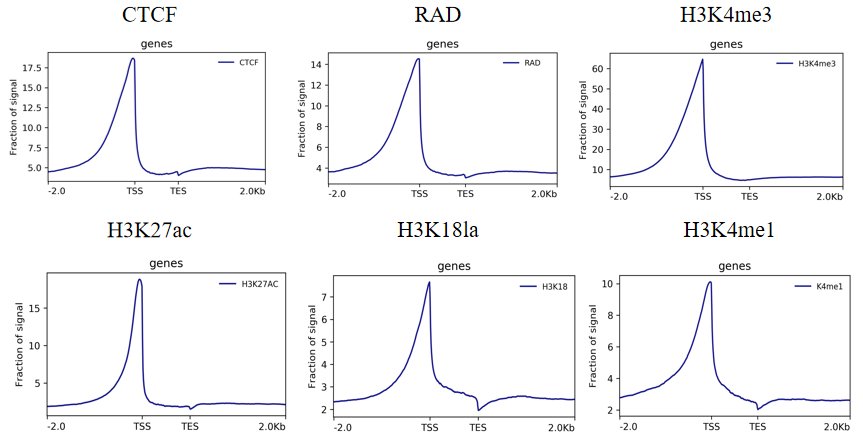
3.reads密度分布图
reads在TSS附近呈现明显的富集。
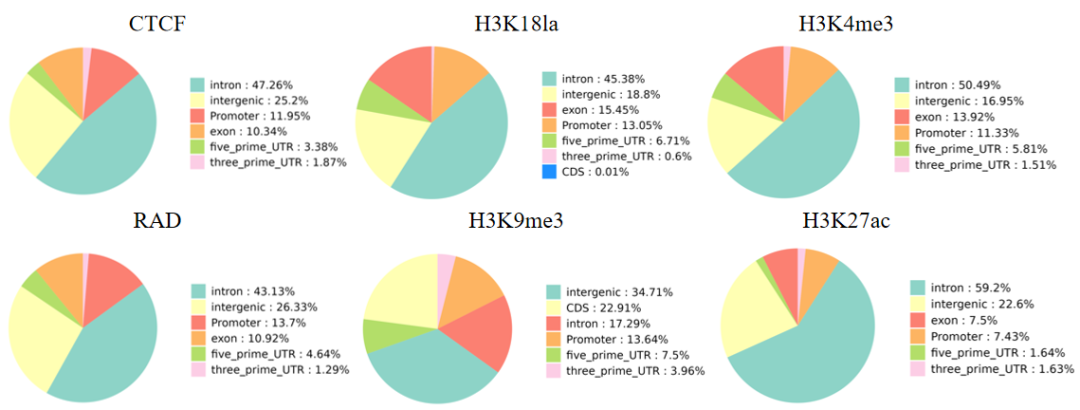
4.功能元件分布图
Peak在基因功能元件上分布。
5.Peak可视化
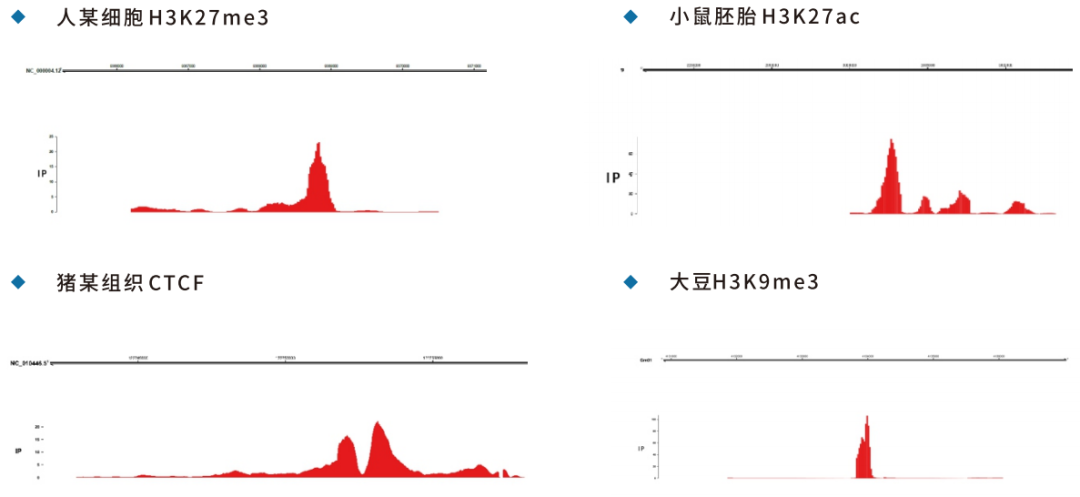
-
参考文献
1. Di X, Xiang L and Jian Z (2023), YAPmediated mechanotransduction in urinary bladder remodeling: Based on RNA-seq and CUT&Tag.Front. Genet. 14:1106927.
2. Xu H, Wu D, Xiao MM, et al(2024), PP2A complex disruptor SET prompts widespread
3. hypertranscription of growth-essential genes in the pancreatic cancer cellsSci. Adv. 10, eadk6633.
4. Zeng J, Chen J, Li M, Zhong C, Liu Z,Wang Y, Li Y, Jiang F, Fang S and Zhong W (2023), Integrated high-throughput analysis identifies super enhancers in metastatic castration-resistant prostate cancer.Front. Pharmacol. 14:1191129.
5. Zhang, Y., Fang, Y., Tang, Y. et al (2022), SMYD5 catalyzes histone H3 lysine 36 trimethylation at promoters. Nat Commun 13, 3190

















 1105
1105

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









