









 本文通过DFT计算探讨了不同过渡金属对α-MoB2的掺杂对氢进化反应(HER)催化活性的影响,发现铁是最优掺杂元素,铁掺杂的MoB2显示出高效且廉价的催化特性。研究还揭示了掺杂剂对反应势垒和表面稳定性的影响,为设计新型HER催化剂提供了理论依据。
本文通过DFT计算探讨了不同过渡金属对α-MoB2的掺杂对氢进化反应(HER)催化活性的影响,发现铁是最优掺杂元素,铁掺杂的MoB2显示出高效且廉价的催化特性。研究还揭示了掺杂剂对反应势垒和表面稳定性的影响,为设计新型HER催化剂提供了理论依据。
 研究背景
研究背景
丰富的过渡金属硼化物正在成为有前途的电化学析氢反应(HER)催化剂,具有取代贵金属的潜力。那些含有类石墨烯(扁平)硼层的材料,如α-MoB2,特别有前途,它们的性能可以通过第二种金属的掺杂进一步提高。捷克马萨里克大学FilipDohnal和帕拉茨基大学Dr.PetrLazar等人采用DFT计算研究了不同过渡金属与α-MoB2取代掺杂后对HER催化活性的影响,以寻找提高HER催化活性的途径。
计算方法
第一原理DFT计算通过VASP进行,使用优化的范德华力vdW-DF2泛函。选取MoB2晶体[0001]晶面,模型采用8个原子层厚,2×2超晶胞。真空层设置为14Å,以避免周期单元之间的可能相互作用,并使用6×6×1的k点网格对布里渊区进行采样,平面波截断能设置为400eV。使用NEB方法进行反应势垒的计算和过渡态识别(Tafel步骤)。
结果与讨论
作者通过DFT计算了MoB2掺杂的各种过渡金属原子(V、Cr、Mn、Fe、Co、Hf、Ta、W、Re、Os)在Mo原子三维框架内的热力学稳定性,以发现哪些金属与母体α-MoB2晶格形成稳定的三元相。通过检查掺杂剂的体积与表面稳定性,以判断掺杂剂是否会在表面分离,从而影响HER的活性。催化活性从氢吸附的微分吉布斯能(ΔGH*)和掺杂剂HER的Tafel步骤的反应势垒来评价。结果表明,掺杂剂对反应势垒和反应途径有不同的影响,铁是最适合MoB2掺杂的元素。 作者考虑在相对较大的掺杂剂浓度Mo0.75X0.25B2下掺杂MoB2,这与最近实验研究中的常见掺杂剂浓度相对应。从表1可以看出对于第三和第五元素周期过渡元素(3d和5d),形成能随着原子序数从V到Co和Hf到Os的增加而逐渐增加。
表1:掺杂Mo0.75X0.25B2的形成能、化学势及晶格参数
3d中V和Cr,5d中Hf和Ta具有负的形成能。因此,这四种元素可以与MoB2形成稳定有序的三元相。3d掺杂元素组成的三元合金是自旋极化的,与之相反由5d元素形成的合金中均未产生磁矩。
体相与表面相比,Fe、Mn、Cr、W、Re和Os掺杂在表面的形成能低于体相,即表面更稳定,V、Co和Ta掺杂出现了相反的趋势,即体相稳定性更好。其中Fe和Mn的形成能由体相的正能量转变为表面的负能量,因此Fe和Mn掺杂的表面更稳定,不易在表面分解成纯相,这对催化剂的长期稳定性很重要。掺杂剂的磁矩在表面处被猝灭,在所有研究的掺杂剂中,只有Cr和Mn掺杂的表面保持磁性。
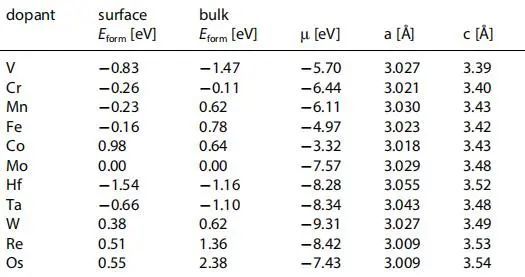
对于掺杂剂扩散到材料体层中的可能性,作者计算了放置在超晶胞板内四个金属层中的每一层中的掺杂剂的形成能(图1)。这些计算是针对形成能接近于零的钨和铁掺杂剂进行的。结果表明,钨和铁掺杂剂最稳定的位置都在表面金属层中。得出这些掺杂剂更倾向于取代表面层中的Mo原子,因此将向Mo端的表面扩散,在表面可以更有效地影响掺杂MoB2的催化活性。
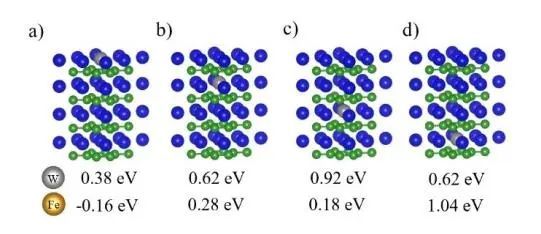
图1 W和Fe掺杂MoB2在各原子层的形成能
析氢反应由电极表面上两次连续的质子电子转移组。反应的第一次转移(Volmer步骤)是氢质子在电催化剂表面的活性位点上的还原。HER目前有两种已知机制:Volmer-Heyrovsky和Volmer-Tafel。在Volmer-Tafel机制中(图2),氢的释放通过两个被吸收的氢原子的重组进行。在Volmer-Heyrovsky机制中,第二次质子-电子转移不是发生在质子的吸附过程中,而是发生在质子与吸附的氢原子的复合和析氢过程中。反应可以用以下方程描述:
Volmer:H3O++e-+*=H2O+H* (1)
Tafel:H*+H*=H2 (2)
Heyrovsky:H3O++e-+H*=H2O+H2 (3)
其中*表示催化剂的活性位点,H*表示吸附在活性位点上的氢原子。

图2 HER的Volmer-Tafel机制示意图
在原始MoB2的[0001]表面上—在一个金属原子的顶部(T)上,在两个相邻金属原子之间的桥位(B)上,以及在三个金属原子(H)之间的空位上,存在三种对称的不等价活性位点。如图3,蓝色表示Mo原子,灰色表示掺杂原子,绿色表示B原子。
图3 掺杂MoB2[0001]表面吸附位点示意图
对于同一周期的掺杂原子,随着原子序数左向右变化,顶部位置的ΔGH*逐渐减小,对于5d掺杂原子这一趋势尤其明显。除了Re和Os掺杂模型将氢吸引到顶部位点,其余氢均优先结合在中空位点上。 作者计算了这些能量如何随着额外吸附的H原子而变化(图4a)。在原始MoB2上,H原子逐渐填充空的中空位点(图4b)。在1ML的覆盖范围内,空心位点被占据(ΔGH*=-0.37eV)。然后,额外的氢必须吸附在Mo原子上方的顶部位置,导致ΔGH*的急剧增加(图4a),覆盖率为1.25ML(ΔGH*=0.29eV)。在掺杂表面上,掺杂剂原子的存在改变了表面位置的填充顺序,并且每种掺杂剂的ΔGH*的覆盖依赖性都会发生不同的变化。在Fe掺杂的表面上,氢在中空位置的吸附率仅为0.5ML。在0.5和1.0之间的覆盖率下,额外的氢填充靠近掺杂剂原子的低对称性位点(图4b)。这种排序在这一组数据中是独特的,并导致其ΔGH*随氢覆盖率呈现出准线性变化关系(图4a)。
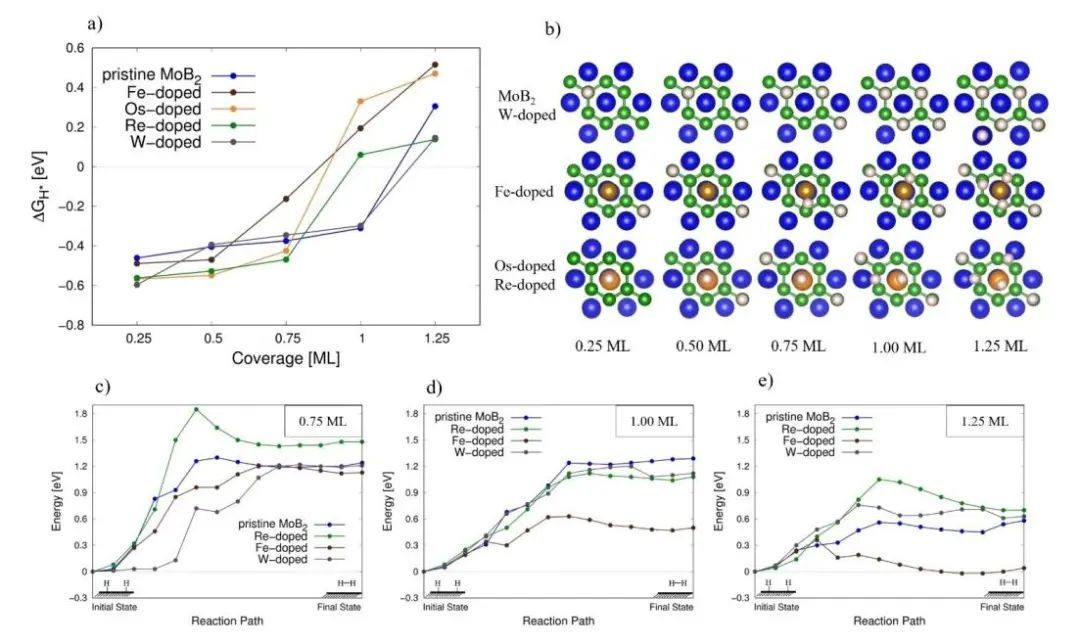
图4 原始和掺杂W、Re、Fe、Os的MoB2模型
在HER的Tafel步骤中作者使用轻推弹性带方法研究这一步骤的反应路径。选择了四个模型:原始MoB2和W、Re、Fe掺杂MoB2,它们代表了初始状态几何形状的三种特征模式(图4b)。对于每个表面,我们考虑ΔGH*接近热中性的三种不同的氢覆盖率:0.75ML、1.0ML和1.25ML(图4c–e)。W掺杂产生的激活势垒与未掺杂MoB2上的激活势垒相似。在0.75ML覆盖率下,Re原子的掺杂导致反应势垒甚至高于原始MoB2上的反应势垒,表明掺杂甚至可能对HER活性产生有害影响。Fe掺杂在1.0ML和1.25ML覆盖范围内都能带来最显著的激活势垒降低。掺杂表面与未掺杂表面上的反应势垒以类似的方式随着氢覆盖率的增加而降低,只是程度不同。
结论与展望
作者通过DFT计算研究了过渡金属对α-MoB2的取代掺杂,寻找作为系统提高HER本征催化活性的途径。计算得出掺杂剂(V、Cr、Mn、Fe、Hf、Ta)的表面能为负,即这些掺杂剂在MoB2晶格内是热力学稳定的。此外,Cr、Mn、Fe和Os掺杂剂优选取代表面层中的Mo原子,因此可以最有效地影响掺杂MoB2的催化活性。对于Fe掺杂的表面,表面上桥位和空位的吸附点的能量非常接近。Fe掺杂对HER的Tafel步骤的激活势垒产生了最强的降低,并且Fe掺杂表面呈现了ΔGH*随氢覆盖率的准线性变化。因此,铁掺杂的MoB2是高效且廉价的HER催化剂的一个重要发展方向。
文献信息
Dohnal, F., & Lazar, P. (2023). Improving the Catalytic Performance of the Hydrogen Evolution Reaction of α‐MoB2 via Rational Doping by Transition Metal Elements. ChemPhysChem, e202200824.


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









