









 山东大学和中科院的研究人员开发出Ir-CoO/Al2O3催化剂,用于光热CO2制取甲烷,表现出128.9mmolgcat-1h-1的高产率。研究通过DFT计算揭示了CO2和H2在Ir-CoO界面的吸附特性,解释了其高效催化机制。
山东大学和中科院的研究人员开发出Ir-CoO/Al2O3催化剂,用于光热CO2制取甲烷,表现出128.9mmolgcat-1h-1的高产率。研究通过DFT计算揭示了CO2和H2在Ir-CoO界面的吸附特性,解释了其高效催化机制。

目前,光热催化CO2加氢是广泛研究的最有前途的方法之一,在温和条件下CO2转化为增值化学品,但是实现理想的转换效率和目标产品选择性仍然具有挑战性。
基于此,山东大学王凤龙教授和中科院山西煤炭化学研究所温晓东研究员等人报道了由Ir/CoAl LDH复合材料衍生制备的Ir-CoO/Al2O3催化剂,用于光热CO2制取甲烷,该催化剂由Ir-CoO系综作为活性中心,均匀地锚定在非晶态Al2O3纳米片上。在250 °C环境压力和可见光照射下,CH4产率达到128.9 mmol gcat-1 h-1,优于大多数报道的金属基催化剂。
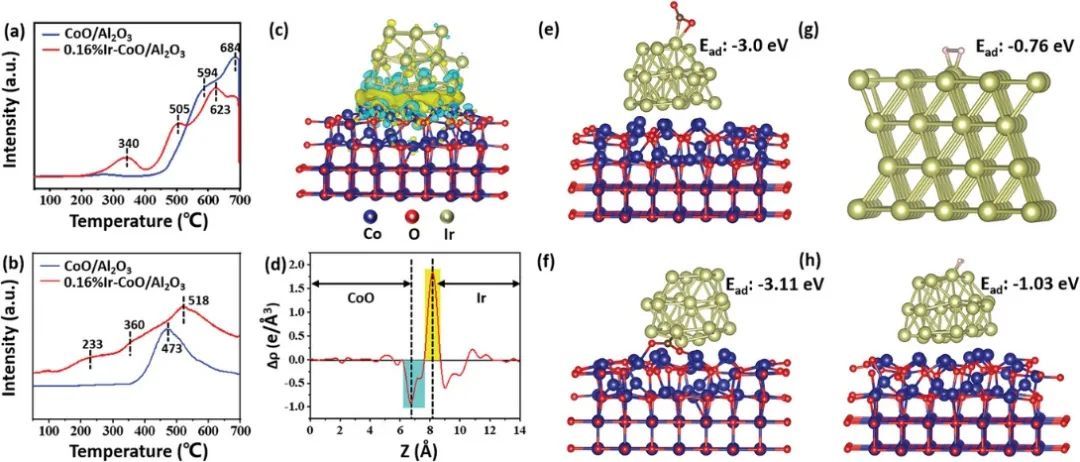
通过DFT计算,作者研究了CO2在Ir表面和Ir-CoO界面上的吸附行为。优化后的CO2在Ir表面和Ir-CoO界面上的吸附几何图发现,Ir面C-O键长从1.16 Å增加到1.22 Å和1.33 Å,键角从180°减小到142.4°;Ir-CoO界面C-O键长分别增大到1.28 Å和1.29 Å,键角减小到127.2°,表明CO2分子在Ir-CoO界面更容易解离。因此,Ir-CoO界面被认为是CO2分子最活跃的吸附位点。
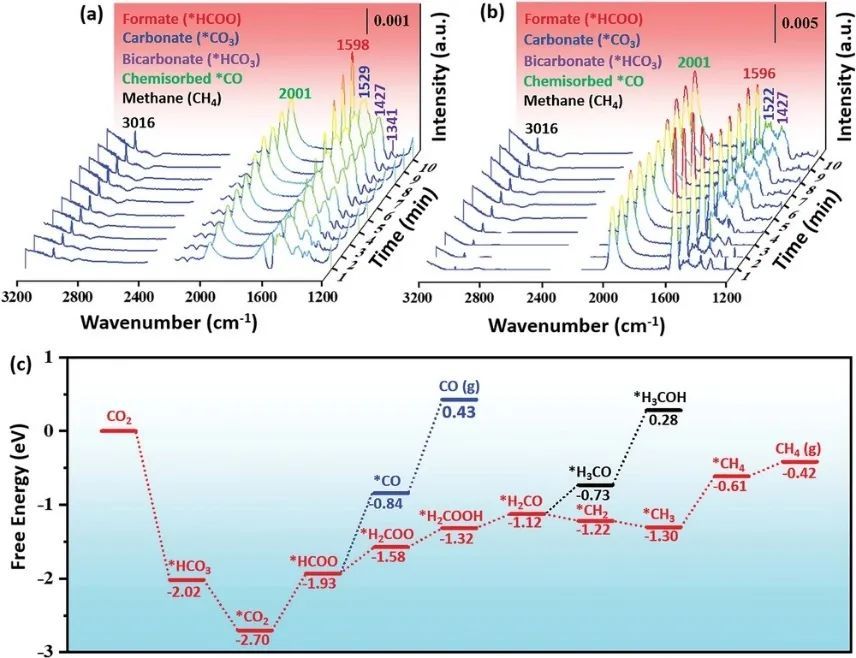
此外,作者还通过DFT计算探讨了H2分子在Ir纳米颗粒表面的吸附行为。通过构造Ir(111)和Ir(111)-CoO的几何图形,分别记为[Ir(111)]和[Ir(111)]-,模拟了带中性电荷和负电荷的Ir表面。H2在[Ir(111)]和[Ir(111)]-表面上的优化几何形状,并模拟了相应的键长和吸附能,表明H2更容易吸附在[Ir(111)]-表面上。 H2分子的σ*反键轨道可以接受来自带负电荷的Ir表面的电子,将使吸附的H2解离成H物种,H物种很容易与吸附的CO2分子反应,从而引发加氢过程。
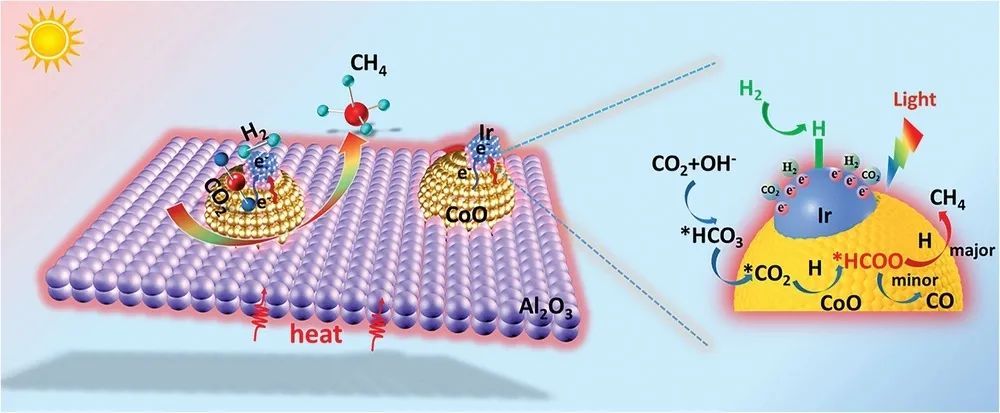
Ir-CoO Active Centers Supported on Porous Al2O3 Nanosheets as Efficient and Durable Photo-Thermal Catalysts for CO2 Conversion. Adv. Sci., 2023.


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









