







GENIE3算法
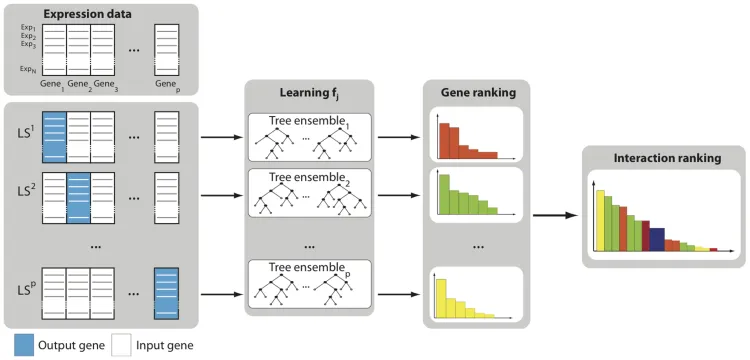
GENIE3 (GEne Network Inference with Ensemble of trees) 是一种基因网络推断算法,用于从基因表达数据中推断出调控网络。GENIE3 的核心思想是通过构建随机森林(或更广泛地讲,树的集合)来预测每个基因的调控因子。具体来说,它使用树模型来评估各个潜在调控因子(其他基因)的表达模式对目标基因表达的影响。这个算法被认为在推断基因调控网络方面具有很高的准确性和鲁棒性。

算法步骤:
目标基因选择:对数据集中的每个基因逐一进行分析,假设该基因为目标基因。
潜在调控因子选择:剩余的所有基因作为潜在的调控因子。
随机森林建模:通过构建一个随机森林模型(或其他树模型)来预测目标基因的表达水平,该模型的输入是潜在调控因子的表达数据。
重要性评分:根据随机森林模型,计算每个调控因子的特征重要性评分(Feature Importance Score),这反映了该基因作为调控因子的重要性。
网络构建:重复上述过程,对于每个目标基因,生成一个基因调控网络。最终网络由所有的特征重要性评分(即基因之间的调控关系)组成。
适用场景:
基因调控网络重建:GENIE3常用于基因表达数据中基因调控关系的推断,特别是在单细胞RNA测序数据和bulk RNA-seq数据中。
生物信息学研究:可以帮助研究人员理解基因之间的调控机制,识别出潜在的关键调控因子和调控通路。
疾病研究:通过识别特定基因的调控网络,可以揭示疾病相关的基因调控机制。
优点:
由于使用了随机森林等集成学习方法,GENIE3对噪声数据具有很强的鲁棒性。
可以适用于不同规模的基因表达数据集,从小规模的实验数据到大规模的高通量测序数据。
缺点:
由于需要对每个基因构建多个随机森林模型,计算量较大,尤其是对于大规模基因表达数据集。
虽然GENIE3能够捕捉非线性关系,但其精度依赖于数据的质量和复杂性,有时可能无法捕捉非常复杂的调控关系。
R语言和Python实现:
在R语言中,可以使用GENIE3包来实现这一算法。 在Python中,可以通过pySCENIC包中的实现来使用GENIE3算法。
分析流程
1、导入矩阵数据和转录因子
rm(list=ls())
library(GENIE3)
load("~/data.Rdata")
exp1_input <- exp1[intersect(degs_1, rownames(exp1)), ]
head(exp1_input)[1:5,1:5]
# GSM3106326 GSM3106328 GSM3106329 GSM3106330 GSM3106332
# NKD1 8.301358 8.475126 8.469785 8.844339 8.669053
# PIEZO2 9.210208 8.769019 8.722026 8.689316 8.001801
# ANKRD36 8.834924 8.628393 8.854510 8.485708 8.475799
# ANKRD36B 9.350684 9.150061 9.273654 8.887777 8.932125
# HMCN1 9.532162 9.362445 10.061496 9.551432 9.162787
# 导入TFs
library(data.table)
tfs <- fread("~/allTFs_hg38_new.txt",header = F)
A1 <- intersect(tfs$V1,rownames(exp1_input))
length(A1)
# [1] 12
需要提前将转录因子和矩阵基因取交集, 因为目前已知的转录因子数量是有限的,而且未必会出现在分析的矩阵中。
2、 GENIE3正式分析-构建共表达网络
# Genes that are used as candidate regulators
regulators_1 <- A1
weightMat_1 <- GENIE3(exp1_input, regulators=regulators_1,
nCores=4, verbose=TRUE,
#K=7, nTrees=50
)
dim(weightMat_1)
# [1] 12 213
限定转录因子的好处在于能够帮使用者提前规定好TFs和靶基因的身份(指定上下级方向)。
3、提取共表达网络数据信息
linkList1 <- getLinkList(weightMat_1,
#reportMax=5, # 这个是提取网络的数量为5
threshold=0.1 # 权重高于0.1
)
dim(linkList1)
# [1] 2544 3
head(linkList1)
# regulatoryGene targetGene weight
# 1 HIVEP2 HIVEP1 0.4859376
# 2 HIF3A BTNL9 0.4226050
# 3 HIVEP1 HIVEP2 0.4178483
# 4 ZNF521 FAM171B 0.3757616
# 5 ZNF189 PKP2 0.3665207
# 6 HIF3A FAM107A 0.3506742
write.csv(linkList1,"linkList1.csv")
接下来就可以放到Cytoscape中进行可视化啦~
参考资料:
1、Inferring regulatory networks from expression data using tree-based methods. PLoS One. 2010 Sep 28;5(9):e12776.
2、 GENIE3:https://bioconductor.org/packages/release/bioc/vignettes/GENIE3/inst/doc/GENIE3.html
注:若对内容有疑惑或者有发现明确错误的朋友,请联系后台(欢迎交流)。更多内容可关注公众号:生信方舟
- END -

















 8266
8266

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









