







SingleR 是一种用于单细胞 RNA 测序(scRNA-seq)数据分析的自动注释工具。它的主要功能是基于参考数据集为未注释的单细胞数据进行细胞类型注释。SingleR 使用了来自已知细胞类型的参考数据集(如公共的参考表达谱数据库)来推断新样本中每个细胞的类型。
SingleR 的工作原理:
-
输入数据:用户需要提供一个待注释的测试数据集(通常是单细胞 RNA 测序的表达矩阵)和一个已知细胞类型注释的参考数据集。
-
参考数据集匹配:SingleR 逐一比较待注释的每个细胞或细胞群与参考数据集中的每个细胞类型的基因表达模式,计算出相似性评分。
-
标签分配:根据与参考细胞类型的相似性,SingleR 将最匹配的细胞类型标签分配给每个测试细胞。
-
置信度计算:SingleR 计算当前预测标签与次优标签之间的差异(如 delta.next),并根据置信度来决定是否保留或修剪预测结果。
-
输出:输出包括初始的预测标签(labels)、修剪后的标签(pruned.labels)、和不同标签之间的置信度差距(delta.next),帮助研究人员理解细胞类型注释的可靠性。
SingleR 的优势:
● 自动化和高效:无需手动设置 marker 基因,SingleR 可以自动从参考数据集中进行学习和预测。
● 灵活性:支持多种参考数据集,包括人类和小鼠的不同细胞类型数据库。
● 准确性:通过置信度修剪,可以减少错误注释的风险。
步骤流程
1、导入
导入处理好的原始数据
rm(list = ls())
library(Seurat)
load("obj.Rdata")
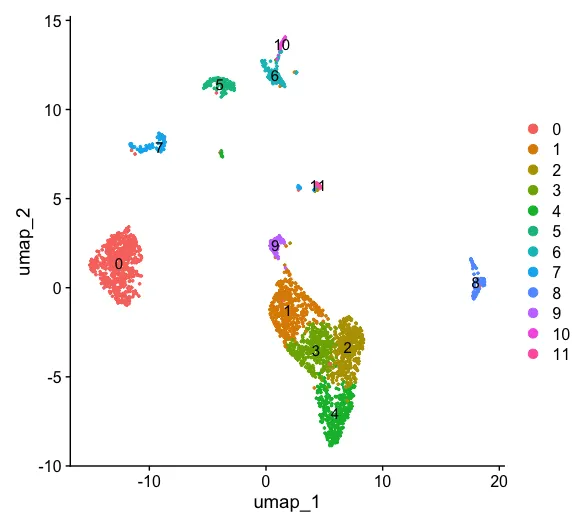
p1 = DimPlot(seu.obj,reduction = "umap",label = T)
p1
一共有12群
2、加载注释文件和R包
library(celldex)
ls("package:celldex")
HumanPrimaryCellAtlas <- HumanPrimaryCellAtlasData()
BlueprintEncode <- BlueprintEncodeData()
MouseRNAseq <- MouseRNAseqData()
ImmGen <- ImmGenData()
DatabaseImmuneCellExpression <- DatabaseImmuneCellExpressionData()
NovershternHematopoietic <- NovershternHematopoieticData()
MonacoImmune <- MonacoImmuneData()
# 其中,以Data结尾的是celldex提供的参考数据,其他的是函数。数据共有7个,除了ImmGenData和MouseRNAseqData是小鼠的,其他都是人类的。
读取HumanPrimaryCellAtlas文件看一下
library(SingleR)
library(BiocParallel)
scRNA = seu.obj
# 提取归一化数据
test <- GetAssayData(scRNA, layer = "data")
#test = scRNA@assays$RNA$data
table(scRNA@active.ident)
# 0 1 2 3 4 5 6 7 8 9 10 11
# 638 476 427 388 354 129 127 126 90 88 31 26
Rename_scRNA <- SingleR(test = test,
ref = HumanPrimaryCellAtlas, # 参考注释文件
labels = HumanPrimaryCellAtlas$label.main, #标注信息
clusters = scRNA@active.ident)
Rename_scRNA$labels
# [1] "B_cell" "T_cells" "T_cells" "T_cells"
# [5] "T_cells" "B_cell" "Endothelial_cells" "Monocyte"
# [9] "Smooth_muscle_cells" "NK_cell" "Endothelial_cells" "T_cells"
Rename_scRNA$delta.next
# [1] 0.066315004 0.389803688 0.968866652 0.777167256 0.841875081 0.381917374 0.106055936
# [8] 0.146110693 0.024163437 0.084589179 0.086608735 0.004900357
Rename_scRNA$pruned.labels
# [1] "B_cell" "T_cells" "T_cells" "T_cells"
# [5] NA "B_cell" "Endothelial_cells" "Monocyte"
# [9] "Smooth_muscle_cells" "NK_cell" "Endothelial_cells" NA
new.cluster.ids <- Rename_scRNA$pruned.labels
names(new.cluster.ids) <- levels(scRNA)
scRNA <- RenameIdents(scRNA,new.cluster.ids)
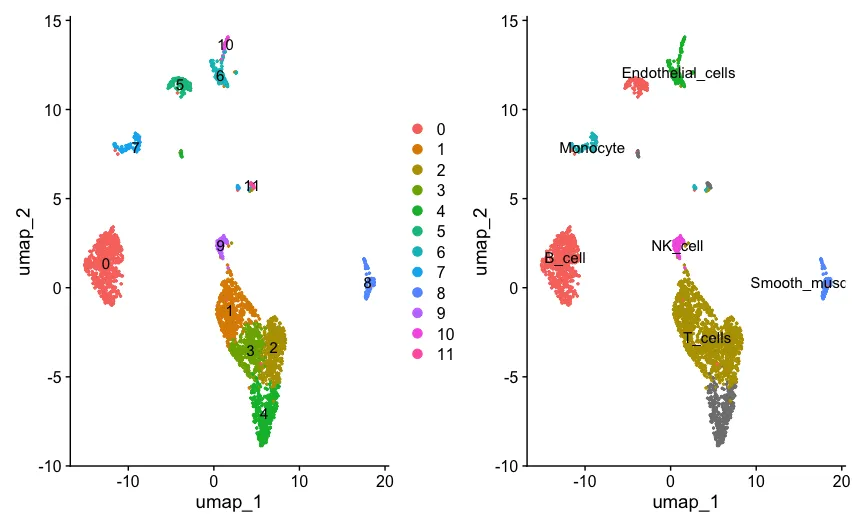
p2 <- DimPlot(scRNA, reduction = "umap",label = T,pt.size = 0.5) + NoLegend()
p1+p2
labels 是 SingleR 方法给每个细胞群或每个细胞分配的标签,表示该细胞的分类结果。这个标签是基于参考数据(如 HumanPrimaryCellAtlas)进行注释的。例如,“T_cells”、“B_cell”等代表细胞类型。
delta.next是每个细胞分配到的第一和第二最匹配标签之间的分数差异。这个值反映了分类结果的置信度。较大的 delta.next 值表示分类的置信度较高,因为第一和第二标签之间的分数差异大;较小的 delta.next 值则表示置信度较低,分类结果可能不太确定。
pruned.labels 是经过修剪的标签结果。当 delta.next 的值过小时,意味着该细胞的分类不够可靠,SingleR 会将这些分类结果设置为 NA,即未分类,以提高分类的准确性。因此,pruned.labels 是去除低置信度分类后的标签,保留了分类结果较为可靠的细胞标签。
但是我们仔细看修建后的结果的第五群,在label中是Tcell,在delta.next中分数也不低!然后在pruned.labels中被NA了~ 让人纳闷。
笔者觉得自动注释只能作为参考,还是要踏踏实实的看文献自行注释~ (求“人/工具”不如求自己啊 hhh)
参考资料:
1、SingleR:https://www.bioconductor.org/packages/release/bioc/html/SingleR.html
2、生信技能树:https://mp.weixin.qq.com/s/GpOxe4WLIrBOjbdH5gfyOQ
致谢:感谢曾老师、小洁老师以及生信技能树团队全体成员。
注:若对内容有疑惑或者有发现明确错误的朋友,请联系后台(欢迎交流)。更多内容可关注公众号:生信方舟
- END -

















 362
362

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









