








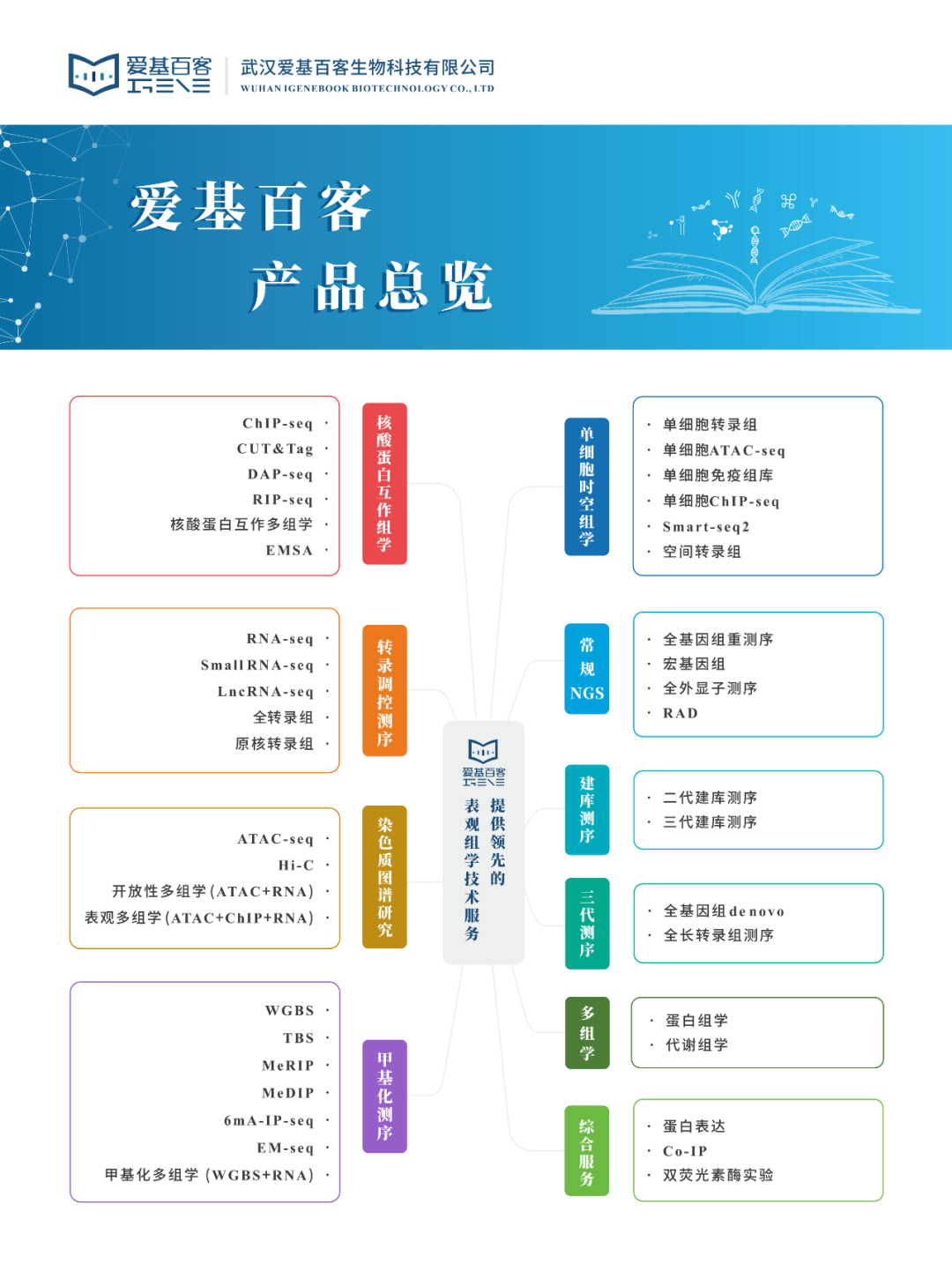
DNA亲和纯化测序技术(DAP-seq)是体外研究DNA与蛋白结合情况的技术,将蛋白质体外表达技术与高通量测序技术相结合,使用体外表达的TF蛋白对裸露的全基因组DNA片段进行孵育,以确定结合序列。这一技术逐渐成为植物转录因子研究中的重要工具。
一、为何选择DAP-seq?
-
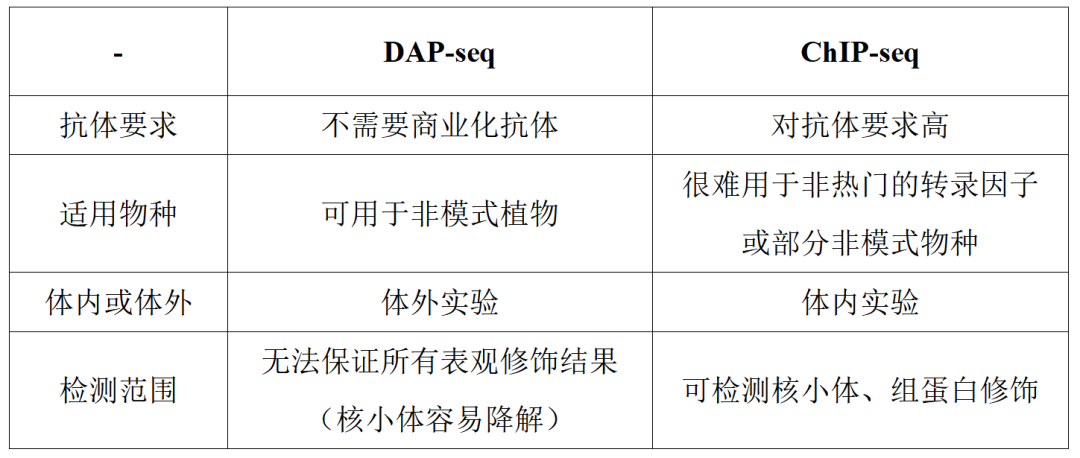
技术优势
-
适用范围
-
抗体难以获得的样本——有些物种(尤其是植物)中的转录因子没有对应的ChIP级别抗体或者没有成熟的转基因体系无法使用标签抗体,而自制抗体周期较长且难度较大;
-
染色质提取困难的样本——有些植物的果实、花、茶叶等糖酚含量较高,提取染色质难度较大;
-
蛋白表达丰度低的样本——植物中的转录因子表达微弱且存在时空表达特性。
二、实验流程概述
1. 前期准备
(1)样本准备:足量样本(详情参考送样指南)、含有目的转录因子的质粒(需提供测序结果)
(2)样本重复:针对同一个转录因子可以不设置重复样本;如果想设置重复样本,建议两到三个,且一次性完成重复实验
(3)分析信息准备:本研究物种匹配的成熟的基因组信息(包括基因组 fa 文件、gff 文件、pep.fa 文件)
备注:理论上植物转录因子都可以做DAP-seq,非植物样本理论上可行,但由于DAP是植物的表达系统,无法保证后续结果。另外,非转录因子的蛋白也不保证后续结果。
2. 具体步骤
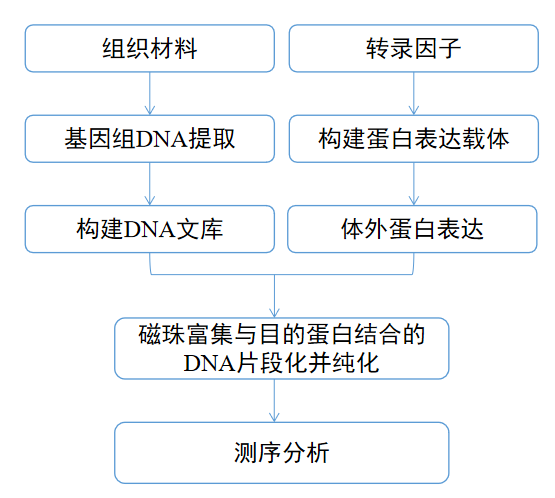
主要步骤包括DNA文库构建、蛋白表达、蛋白与文库的结合反应、文库PCR加接头及定量检测、上机测序、生信分析等。
(1)组织材料的全基因组 DNA 提取,并构建 DNA文库;
(2)构建转录因子的Halo-tag体外表达质粒,利用麦胚系统进行蛋白表达;
(3)混合蛋白与文库,磁珠富集与目的蛋白结合的 DNA 片段;
(4)多次洗涤,除去非特异结合的染色质,并纯化复合体;
(5)纯化 DNA片段,测序分析。
简易流程如下图所示:
3. 分析流程

(1)首先会对Raw data进行质控以及数据过滤,包括接头的过滤以及低质量数据的过滤,并且对过滤完成的clean data进行质控;
(2)利用clean data进行参考基因组序列比对,也就是看一下测序获得的这些reads是位于基因组上的哪些位置;
(3)比对完成后会生成bam文件,利用软件对bam文件进行call peak,简单来看就是看在哪些位置有reads的显著性堆积,这个位置能体现出研究的转录因子结合的位置在哪里;
(4)得到peak后会对peak进行基本分析,包括peak的数量、长度、在基因组上的分布、在基因功能元件上的分布等;
(5)对peak关联到的基因进行GO、KEGG注释和富集分析,以此了解这些基因的功能等。还会对关联基因进行动植物转录因子预测,也就是这些基因可能属于哪种转录因子。
(6)最后一个重要的内容是motif分析。Motif序列可能就是这个转录因子能去结合的特征。进行motif预测便于了解转录因子结合特性以及后续的验证。
(分析报告会对每一部分进行详细的展示,感兴趣的老师可以联系当地销售获取报告demo)
三、项目文章速览
文 章 一

发表期刊:The Plant Journal(IF2022=7.1)
技术路线:
◆ 主要内容 ◆
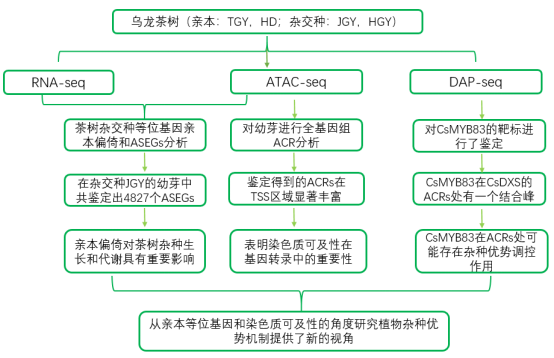
在本研究中,作者对中国栽培面积最大的人工乌龙茶进行了等位基因和染色质可及性水平分析。作者首先找到了杂种优势形成的原因,然后利用ATAC-seq发现了杂交种的ACRs明显高于亲本,这可能使杂交种的基因转录活性更广泛、更强。杂交种中显著增加的ACR有助于调节ASEG的表达,从而影响杂种优势代谢产物的形成。此外,作者还通过DAP-seq验证了速率限制酶CsDXS启动子ACR中CsMYB85的结合motif。这些结果为从亲本等位基因和染色质可及性的角度研究植物杂种优势机制提供了新的视角。然而,该研究无法识别等位基因特异性ACR并分析其与ASEGs的关系。因此,后续研究可以通过构建多个杂交种的单倍型解析基因组,更精确地探索杂种优势形成的等位基因调控。
详细解读:项目文章 | The Plant Journal & DAP-seq助力揭示乌龙茶树杂种优势形成的分子机制
文 章 二

发表期刊:Plant Cell(IF2023=10.676)
技术路线:
◆ 主要内容 ◆
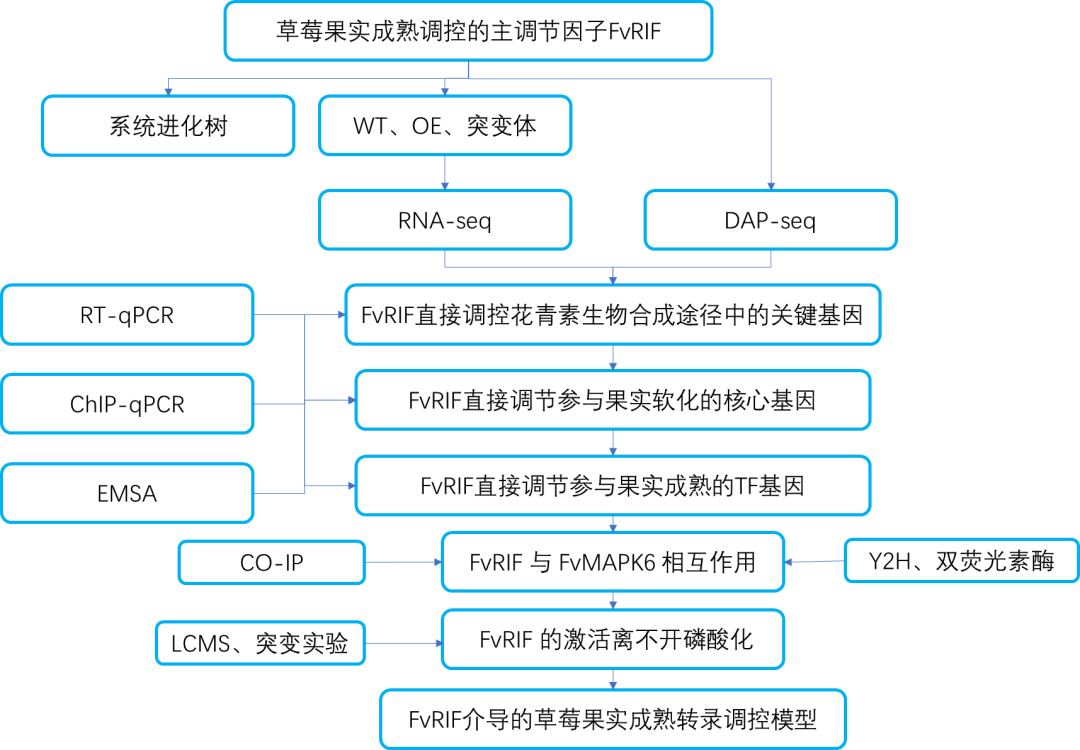
研究发现二倍体草莓Fragaria vesca中的FvRIF是控制果实成熟的关键调节因子,FvRIF的敲除突变导致果实不能成熟发育。DAP-seq与转录组结合表明,2080个基因是FvRIF介导的调控的直接靶点,包括与果实成熟的各个方面相关的基因。研究表明,FvRIF通过直接调节相关核心基因来调节花青素生物合成和果实成熟。此外,还证明FvRIF与MAP激酶6(FvMAPK6)相互作用并作为其底物,MAP激酶6通过在Thr-310磷酸化FvRIF来调节FvRIF。研究结果揭示了FvRIF介导的转录调控网络在控制草莓果实成熟中的作用,并强调了磷酸化修饰对FvRIF成熟活性的生理意义。
详细解读:项目文章| Plant Cell&DAP-seq解析草莓NAC转录因子FvRIF的调控网络
爱基百客专注于表观组学服务领域多年,提供从方案设计、样本制备、测序、生信分析以及后期验证一站式服务。针对DAP-seq技术,我们已具备多个物种的相关项目经验,更多细节请联系区域销售工程师。
















 994
994

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









