






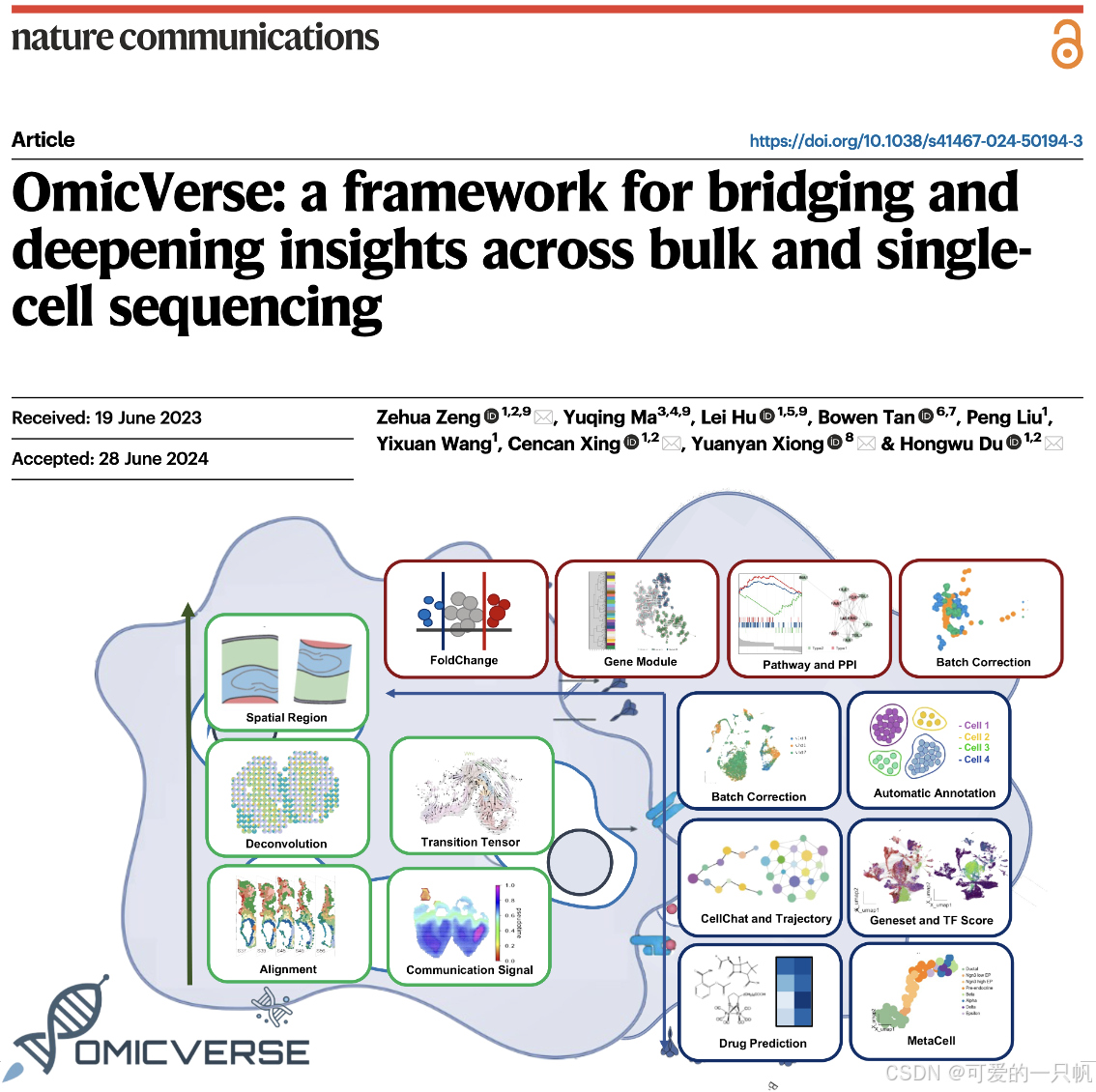
OmicVerse 是一个集成的 Python 框架,旨在简化和增强批量 RNA 测序(Bulk RNA-seq)和单细胞 RNA 测序(scRNA-seq)的分析。它通过整合多种分析工具,为用户提供一致且用户友好的界面,使得在一个编程环境中即可进行全面的转录组分析。
OmicVerse 为批量 RNA-seq 数据提供全面的分析平台
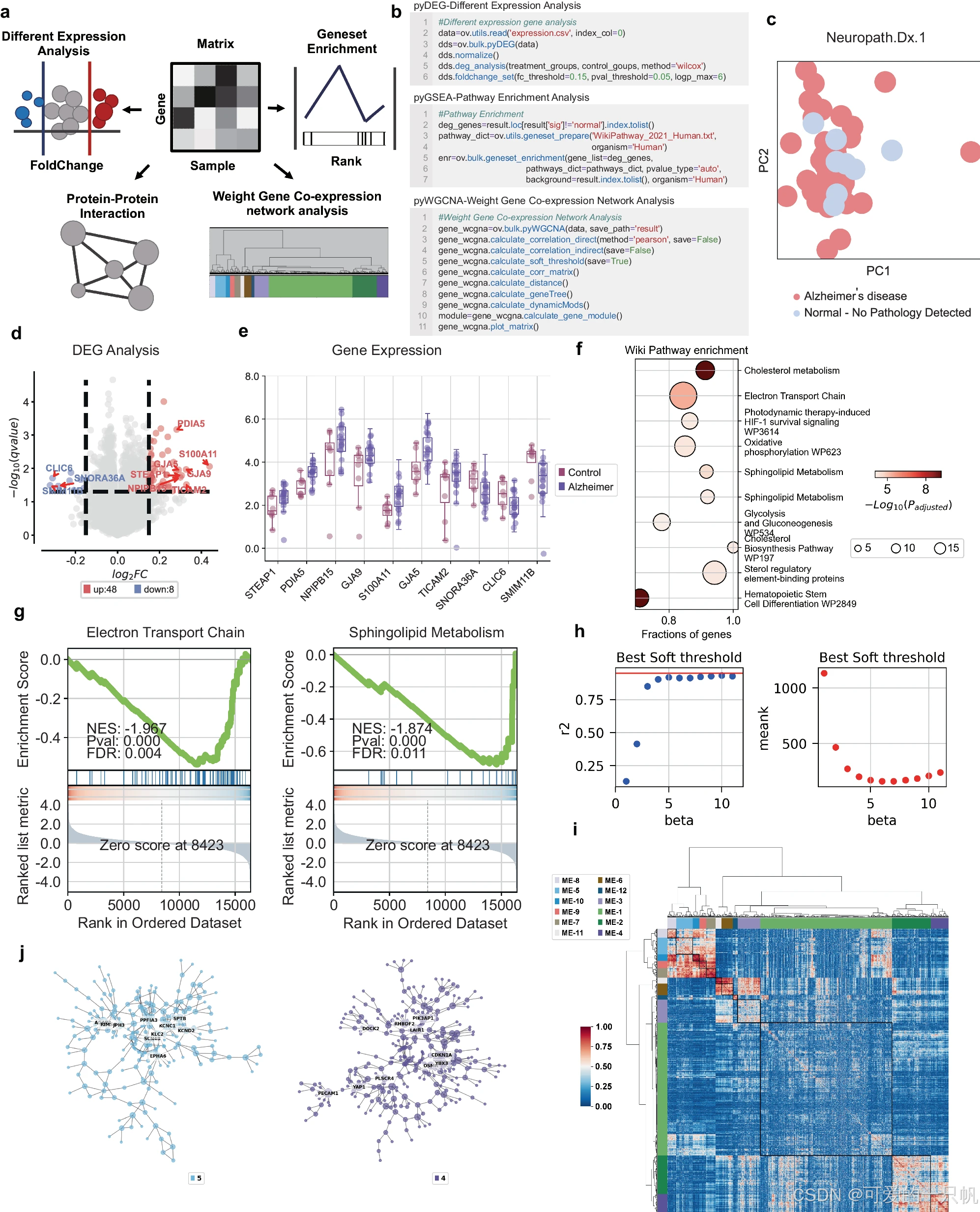
OmicVerse 为单细胞 RNA-Seq 分析提供了一个多功能的多方面框架

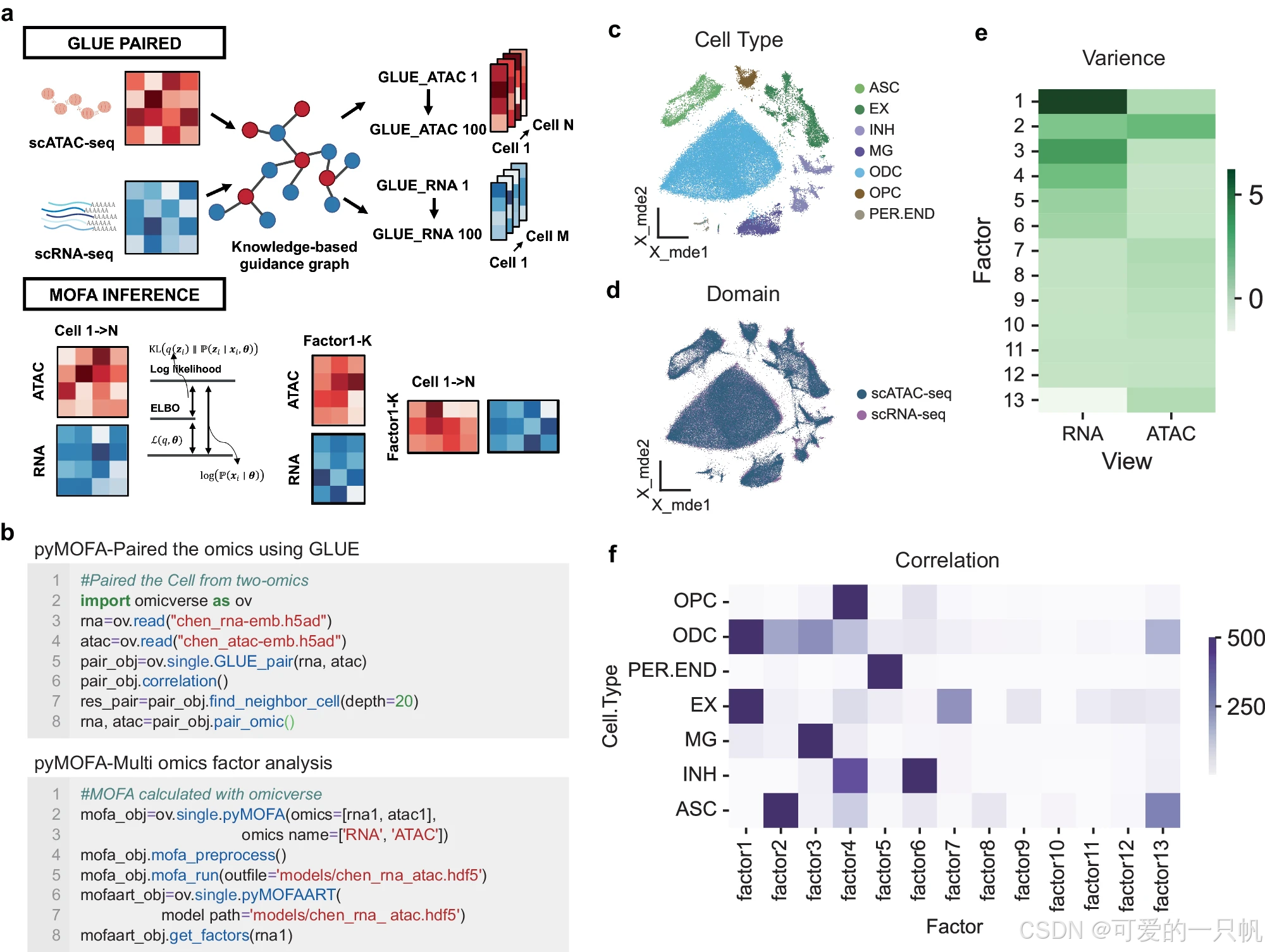
OmicVerse 利用 MOFA 和 GLUE 集成多组学数据分析
主要功能
1. Bulk RNA-seq 分析:
• 提供多样本集成、批量效应校正、差异基因表达分析、基因集富集分析、蛋白质相互作用网络构建、基因共表达模块识别以及 TCGA 数据库预处理等全面功能。
2. 单细胞 RNA-seq 分析:
• 支持多样本质量控制、批量效应去除与集成、自动化细胞类型注释(支持多个数据库)、迁移注释、细胞类型和基因集富集分析、发育轨迹重建、元细胞识别、细胞相互作用网络分析以及药物反应预测。
• 提供 scATAC-seq 集成和多组学分析功能,与 RNA-seq 密切关联。
3. Bulk RNA-seq 到 scRNA-seq 转换:
• 增强了 Bulk RNA-seq 的去卷积、细胞比例估计、scRNA-seq 数据插值以及恢复单细胞数据中的发育轨迹,成为从批量到单细胞 RNA-seq 转换的关键桥梁。
OmicVerse 独特地结合了变分自编码器和图神经网络,以 BulkTrajBlend 框架来解决单细胞数据中的“遗漏”问题,通过从 Bulk RNA-seq 中去卷单细胞数据,精确重构细胞发育轨迹。此外,OmicVerse 通过一个专门的分析对象来简化每个组学层次的分析,确保用户体验的直观性。
参考文献:Nature Communications volume 15, Article number: 5983 (2024)

















 1450
1450

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









