






生信碱移
seismic性状关联细胞鉴定
全基因组关联研究(GWAS)在识别与人类疾病和表型特征相关的遗传变异方面取得了显著成果。与此同时,单细胞RNA测序(scRNA-seq)的兴起使研究人员能够精确分析各类细胞的基因表达。一些研究表明,整合GWAS和scRNA-seq可以精确定位与性状相关的细胞类型,将GWAS中发现的变异与特定基因关联,并通过scRNA-seq分析其细胞类型特异性。
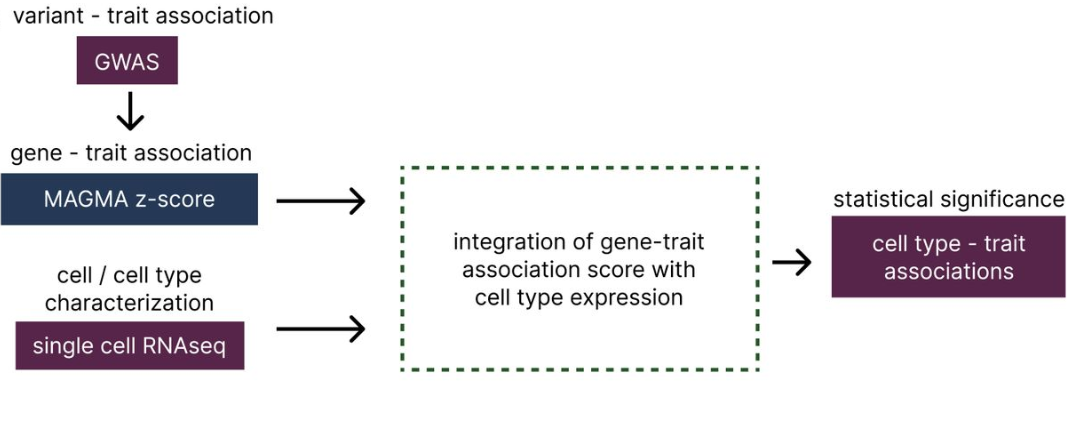
▲ 当前的细胞类型-性状关联方法:通过整合单细胞表达数据与来自MAGMA的基因-性状关联,以统计显著性为依据优先识别细胞类型-性状关联。
目前已有多种计算方法将scRNA-seq参考图谱与GWAS中识别的单核苷酸多态性(SNPs)相结合,以识别与性状相关的细胞类型。主流方法多使用MAGMA处理连锁不平衡问题,并用于预测细胞类型与性状的关系。然而,这些方法存在一些技术局限,包括对基因数量、细胞类型标志物数量的阈值设定过于依赖,忽略了基因表达的变异性,以及处理大规模单细胞数据的扩展性不足。此外,这些方法通常仅输出统计显著性,未进一步解析各基因在细胞类型-性状关联中的具体贡献,限制了研究的深入。

▲ DOI: 10.1101/2024.05.04.592534。seismic与今年5月份上线,尽管仍处于预印本阶段,但是考虑到它的通讯作者在各种顶刊上发表过不少研究,所以还是具有一定学习使用价值。
为此,来自美国莱斯大学的研究人员提出了一款算法框架seismic,该方法能够稳健且高效地发现细胞类型与性状的关联,并同时识别出驱动每种关联的具体基因和生物过程。seismic通过引入考虑基因表达变异的细胞类型特异性评分,避免了对性状或细胞类型关联的任意阈值选择。通过在模拟数据和真实数据上应用seismic和比较现有的基于MAGMA的细胞类型关联方法,作者证明了seismic的校准、效率和分析能力。
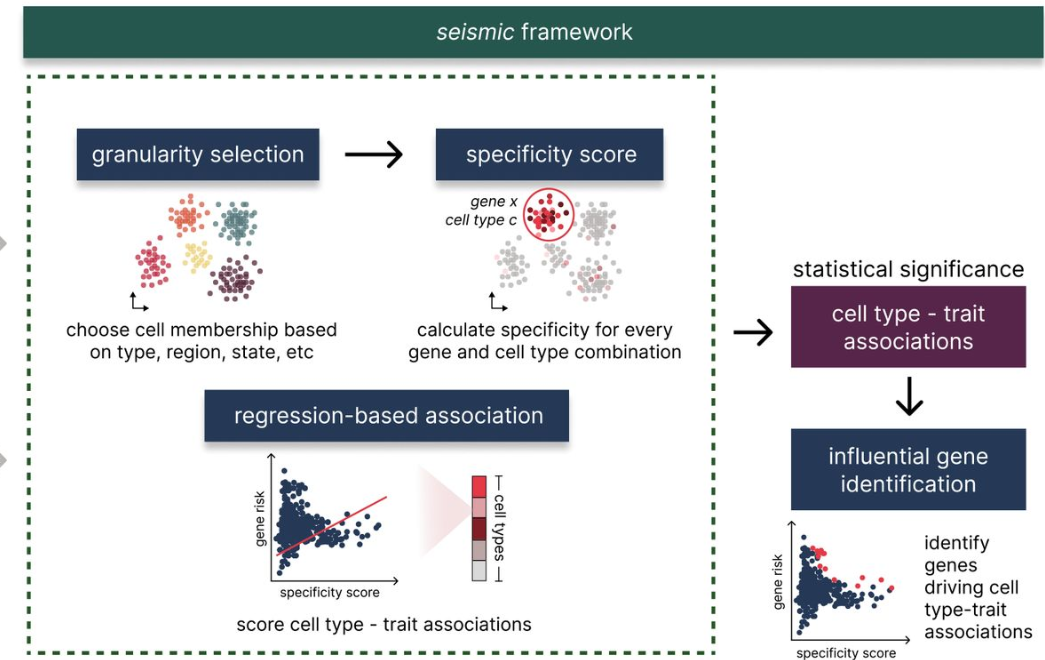
▲ seismic使用MAGMA通过GWAS汇总结果获得基因层面的性状关联数据即z-score;同时利用scRNA-seq数据表征不同细胞类型的基因表达特异性。接着通过回归分析,将基因特异性得分与GWAS z-score结合,评估细胞类型与性状的关联性,并生成显著性结果。seismic框架的亮点在于其可以识别驱动这些关联的关键基因,量化关键基因对细胞类型-性状关联的贡献,用于揭示潜在的生物学机制。
阅读下方链接查看更多内容:
-
https://github.com/ylaboratory/seismic
01.输入数据与R包安装
seismic需要以下数据作为输入:
-
单细胞数据以SingleCellExperiment格式提供,如果拥有seurat对象,可以使用以下代码转换:
# 将seurat对象af转换成SingleCellExperiment格式
af <- Seurat::as.SingleCellExperiment(af)
-
使用MAGMA软件,基于GWAS结果
gwas_results.txt生成的基因水平表型关联结果:
magma \
--bfile 1000G_EUR_Phase3_plink \
--pval gwas_results.txt use=SNP,P ncol=N \ # 指定GWAS结果文件 'gwas_results.txt',使用SNP列作为SNP标识,P列作为p值,N列表示样本数量
--gene-annot 1000G_EUR_Phase3.genes.annot \ # 指定基因注释文件,将SNP映射到基因
--out gwas_gene_results
当然,本文将使用seismic提供的示例文件。在此之前,使用以下代码安装并引用该包:
#devtools::install_github("ylaboratory/seismic")
library(seismicGWAS)
02.示例运行
①示例数据展示。seismicGWAS包含了一个预加载的小型单细胞RNA测序数据集,以SingleCellExperiment对象tmfacs_sce_small的形式提供:
library(seismicGWAS)
tmfacs_sce_small
#class: SingleCellExperiment
#dim: 23341 4000
#metadata(0):
#assays(1): logcounts
#rownames(23341): 0610005C13Rik 0610007C21Rik ... l7Rn6 zsGreen-transgene
#rowData names(1): symbol
#colnames(4000): M4.MAA000844.3_10_M.1.1 A18.MAA000388.3_11_M.1.1 ... M5.MAA000910.3_10_M.1.1 L2.MAA000563.3_10_M.1.1
#colData names(17): nReads orig.ident ... sizeFactor cluster_name
#reducedDimNames(0):
#mainExpName: RNA
#altExpNames(0):
还包括基于2型糖尿病GWAS结果生成的MAGMA示例文件:
head(t2d_magma)
# GENE CHR START STOP NSNPS NPARAM N ZSTAT P
#1 254173 1 1074286 1143315 21 3 572082 1.75840 0.039337
#2 8784 1 1128888 1177163 6 1 572870 1.93210 0.026671
#3 55210 1 1412523 1480067 9 1 569925 0.24436 0.403470
#4 339453 1 1460158 1510740 37 2 571069 0.14879 0.440860
#5 29101 1 1467053 1545262 39 2 571016 0.15608 0.437990
#6 9906 1 1646277 1712438 16 2 576212 -0.48124 0.684830
②计算细胞类型特异性得分。seismic的第一步是计算细胞类型特异性得分,这将用于后续的细胞类型与性状的关联计算以及关键基因分析。使用示例SingleCellExperiment对象和数据中包含的细胞类型标签列cluster_name:
tmfacs_sscore <- calc_specificity(tmfacs_sce_small, ct_label_col='cluster_name')
head(tmfacs_sscore[, 1:3])
# Bladder.Bladder mesenchymal cell Bladder.Luminal bladder epithelial cell Brain_Myeloid.microglial cell
#0610005C13Rik 0.00000000 0.000000e+00 3.639054e-19
#0610007C21Rik 0.05171198 5.416472e-02 3.768650e-02
#0610007L01Rik 0.05053099 5.907819e-02 3.409995e-07
#0610007N19Rik 0.10618537 4.726793e-05 4.422689e-185
#0610007P08Rik 0.04881961 6.890189e-02 2.758088e-08
#0610007P14Rik 0.04976406 6.196749e-02 2.225707e-17
tmfacs_sce_small中的原始单细胞实验数据是小鼠物种的。为了与人类GWAS性状相关联,需要转换基因ID。seismicGWAS提供了一个函数translate_gene_ids,方便地将基因名称进行转换。这里可以将小鼠基因ID转换为人类的Entrez ID,以匹配MAGMA数据:
tmfacs_sscore_hsa <- translate_gene_ids(tmfacs_sscore, from='mmu_symbol')
head(tmfacs_sscore_hsa[,1:3])
# Bladder.Bladder mesenchymal cell Bladder.Luminal bladder epithelial cell Brain_Myeloid.microglial cell
#81932 2.542912e-04 0.01671191 7.503793e-12
#81577 5.003187e-04 0.01487892 3.645070e-10
#8614 9.234388e-02 0.02736780 0.000000e+00
#256281 3.213228e-07 0.08150739 7.970981e-07
#221687 0.000000e+00 0.00000000 0.000000e+00
#346389 0.000000e+00 0.17272120 0.000000e+00
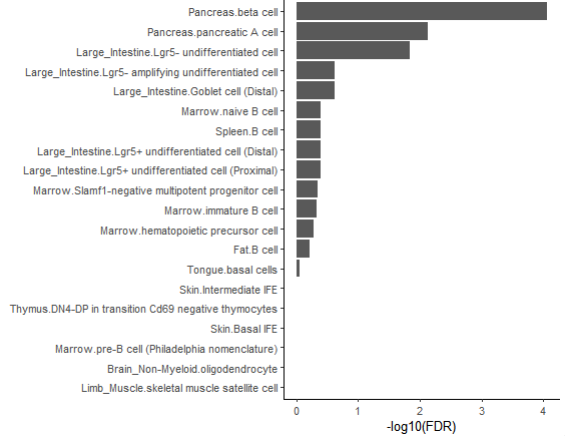
③计算细胞类型与性状的关联。计算2型糖尿病数据的细胞类型与性状关联,并绘制前20个关联细胞的图表。默认图表显示FDR校正后的p值,使用-log标度绘制,因此关联越强的结果在箱线图的顶部:
# 计算单细胞数据中与2型糖尿病性状关联的细胞类型
t2d <- get_ct_trait_associations(tmfacs_sscore_hsa, t2d_magma)
plot_top_associations(t2d, limit = 20)
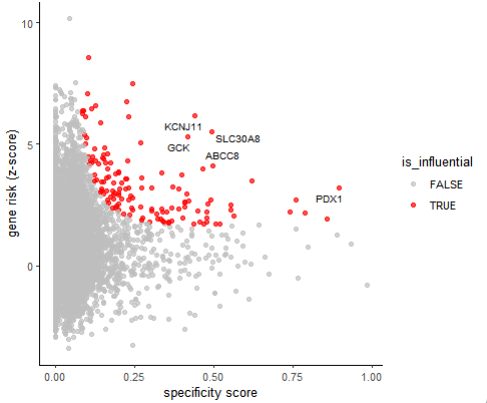
④关键基因分析。借助seismicGWAS,还可以分析驱动特定细胞类型与性状关联的基因,此过程称为关键基因分析。这里,可以选择一个特定的细胞类型(本文选择胰腺β细胞)和一个性状(本文是2型糖尿病)。需要注意的是,关键基因计算仅对在前述细胞类型与性状关联中显著的细胞类型和性状有意义:
# 计算关键基因
ct <- "Pancreas.beta cell" # 选择胰腺β细胞
t2d_beta_inf_genes <- find_inf_genes(ct, tmfacs_sscore_hsa, t2d_magma)
head(t2d_beta_inf_genes)
注意,还需要使用biomaRt包将Entrez ID转换为基因名称,更好的解释结果:
library(biomaRt)
# 转换基因名称
ensembl <- useMart("ensembl", dataset = "hsapiens_gene_ensembl")
hsa.map <- getBM(attributes = c('entrezgene_id', 'hgnc_symbol'),
filters = 'entrezgene_id',
values = t2d_beta_inf_genes$gene,
mart = ensembl)
hsa.map$entrezgene_id <- as.character(hsa.map$entrezgene_id)
t2d_beta_inf_genes <- merge(t2d_beta_inf_genes, hsa.map,
by.x='gene', by.y='entrezgene_id')
# 可视化,num_labels指定标记名称的基因数目
plot_inf_genes(t2d_beta_inf_genes,
gene_col = 'hgnc_symbol',
num_labels = 5)
简单分享到这里
欢迎各位老哥老姐关

















 2729
2729

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









