









- 二代个体重测序
- 1.1 个体重测序部分基本概念
- 个体重测序:对已知基因组序列的物种进行全基因组测序,将不同样品的测序reads与参考基因组进行比对 (不做组装),从而得到个体与参考基因组和个体之间的在基因组序列上变异。
- 文库插入片段:基因组DNA进行片段化处理后,会根据分析需求选择特定长度的DNA片段进行建库,构建到文库中的片 段即为插入片段,重测序文库默认350bp,还可构建270bp、500bp文库。
- 测序深度
- 测序深度:实际测序得到的碱基总量(bp)与基因组 大小的比值,测序带来的错误率或假阳性结果会随着 测序深度的提升而下降。
- 基因组上不同位置测序深度分布不均一,这与基因组 的序列特征和测序深度有关。
- 测序覆盖度:比对到基因组的碱基覆盖基因组的比例,测序 覆盖度是反应测序随机性的一个指标。
- 随测序深度增加,基因组覆盖度也逐渐增加
- 测序深度:实际测序得到的碱基总量(bp)与基因组 大小的比值,测序带来的错误率或假阳性结果会随着 测序深度的提升而下降。
- 1.2 个体重测序研究目的
- 目的:检测样品测序样品间或者样品与参考基因组之间 DNA 变异信息,开发特异分子标记用于后续高级分析,个体重测序是其他高级分析的基础。
- 个体重测序的前提条件是物种必须有 较为完善的参考基因组。
- DNA变异类型
- 单核苷酸多态性single nucleotide polymorphism, SNP
- 小插入缺失(小于50bp) Small Insert and deletion, indel
- 结构变异 Structural Variation,
- SV 拷贝数变异 Copy Number Variations,CNV
- 1.3 个体重测序分析原理
- SNP检测
- 纯合/杂合SNP
- Small indel检测
- 插入/缺失
- SNP检测
- 1.4 个体重测序分析内容
- 分析流程图
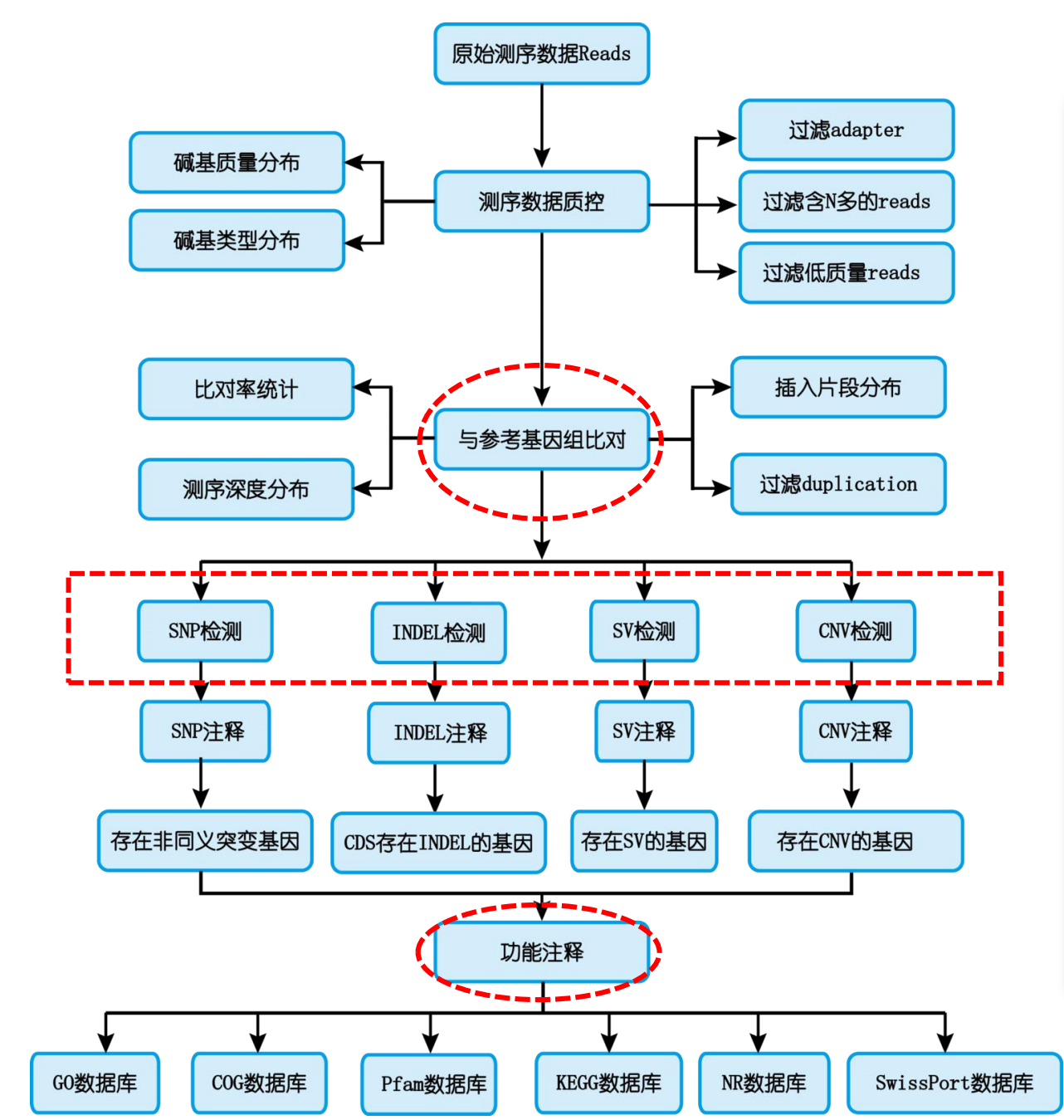
- 与参考基因组比对
- 重测序数据使用BWA软件将二代测序短reads与参考基因组进行比对,即可得到Clean Reads在参考基因组上的位置,统计各样品的测序 深度、基因组覆盖度等信息,并进行变异的检测。
- 比对效率:可以mapping到参考基因组上的Clean Reads占总的Clean Reads数比例,如果参考基因组选择合适,且相关实验过程不存在污 染,测序reads的比对率会高于70%。
- 影响比对率的高低受测序物种与参考基因组亲缘关系远近、参考基因组组装质量高低及reads测序质量有关,物种越近缘、参考基因组 组装越完整、测序reads质量越高,则可以定位到参考基因组的reads也越多,比对效率越高。
- SNP、small InDel检测注释
- SNP、 small InDel的检测主要使用GATK软件,通过多重检测及过滤,最终将得到的变异信息存储在VCF文件中,VCF文 件记录了变异信息在基因组上的位置、碱基类型组成、测序深度等信息,是极为重要的中间文件。
- 样品之间SNP、small InDel的检测是通过将单个样品的VCF合并后得到的,它记录了所有样品之间变异信息,是后续进行 样品间差异比较的基础。
- SNP、 small InDel的注释使用的是SnpEff软件,注释主要分为两部分,一是变异的位置注释,二是功能注释。
- 位置注释即根据变异在基因组上发生的位置,给出变异位点与临近基因的空间关系。
- 功能注释则是指变异发生在基因的CDS区时,可能对基因功能带来影响,并评估不同位置变异的影响力大小。
- 分析流程图
- 1.5 个体重测序测序深度推荐
- 测序深度即影响基因组覆盖度和SNP检出率,如要全 面并准确地检出SNP,推荐测序深度在30x以上 •
- 如需分析SV、CNV等复杂变异,建议测序深度在30x 以上
- 根据不同高级分析内容,测序深度推荐也差异较大, 遗传图谱子代样品推荐1x-5x,GWAS、遗传进化单个 样品推荐5x-10x
- 1.6 个体重测序成功案例
- 水稻亚种基因组测序与重组群体遗传分析揭示地域性产量 与质量相关的遗传位点
- 白菜重测序-遗传进化
- 1.1 个体重测序部分基本概念
- 三代个体重测序
- 2.1 ONT测序平台介绍
- sanger
- illumina
- pb
- 2.2 ONT测序原理
- ONT测序是独一无二的基于电 信号识别碱基序列的三代测序 技术。
- DNA/RNA上不同碱基化学性质存在差异,当单链分子通 过纳米孔通道时,碱基造成的阻碍大小不一,因此会形成 特征性离子电流变化信号。
- 通过对这些信号进行实时检测,即可获得相应碱基类型, 完成测序。
- 目前通过“递归神经网络(Recurrent Neural Network)”的 复杂算法对碱基进行判读,其判定过程相较于荧光信号判 读更为复杂。
- 2.3 ONT测序优势
- 长读长:理论上Nanopore技术的测序平均读长能够达到几十到上百 Kb,最长读长能达到2 Mb以上级 别
- 利于结构变异(拷贝数变异CNV、插入、缺失、易位和倒位)及全长转录本定量及可变剪切检测等;
- 低成本:相比其他三代测序技术,ONT测序样本处理极其简单,无需DNA聚合酶、连接酶和dNTPs, 测序价格低;
- 可不进行PCR扩增,避免二代测序中PCR扩增可能引入的错误;
- 可直接跨越串联重复序列、高GC/AT序列、高度多态性区域、高度同源区域等 特殊区域,检测能力大大优于二代测序
- 可直接读取碱基修饰信息,如甲基化修饰5mC、6mA等,无须像二代测序需要 经过重硫酸盐转化或者免疫沉淀富集实验。
- 2.4 三代个体重测序研究目的
- 分析分析SV(大片段的插入、 缺失)
- SV与人类疾病和动植物的性状变异、遗传进化密切相关
- a. 稀有且相同的一些结构性变异往往和疾病的发生相互关联甚至还是其直接的 致病诱因,比如自闭症、肥胖症、精神分裂症和癌症等都与SVs相关;
- b. CNV覆盖了人类基因组29.74%的序列(SNP仅覆盖1%左右),20%左右的 基因受CNV变异影响;
- c. 研究发现,基因组上的SVs比起SNP而言,更能代表人类群体的多样性特征;
- d. 在植物上面,SV与许多表型变异、生物胁迫/非生物胁迫相关。
- 二代分析SV主要缺点:
- a. 文库大小、检测方法与软件对结果影响比较大,各个方法之间的重合率低;
- b. 读长短,无法准确检测比较大的结构变异和比较复杂的区域内的结构变异,无法准确判断SV断点。
- 三代分析SV的优势:
- a. 三代读长长,平均10-12kb,甚至更长,而SV变异多数集中在50bp-5kb,少数可达到5kb以上;能跨过基因组中大多数SV;
- b. 可以跨过基因组中比较复杂的区域,提供准确的SV检测结果,准确判断SV断点。
- SV与人类疾病和动植物的性状变异、遗传进化密切相关
- 检测外源片段插入
- 外源片段插入(如T-DNA)是创制突变体的有效途径,也是研究突变基因功能常用方式。
- 常规实验方法检测插入片段操作复杂、准确性较差,如Tail-PCR;而二代测序进行插入片段检测时,不能准确找到重组 断点,有时可能因为插入位置的序列特征,根本就不能找到插入位置。
- 三代测序以其长读长的优势,可以轻松跨过外源片段插入区域,还能准确鉴定插入断点。
- 分析分析SV(大片段的插入、 缺失)
- 2.5 三代个体重测序分析原理
- 2.6 三代个体重测序测序指标
- 测序平台:Nanopore
- 文库大小:8 Kb
- 测序深度:15x-20x以上
- 在15x-20x,大片段插入、缺失检出率基本可以 达到80%左右。
- 2.7 产品定位(产品适用场景)
- 前提:有比较完整的参考基因组 •
- 适用情况:
- a. 突变体材料与野生型材料的变异检测(每种类型1个个体)
- d. 转基因事件检测与插入片段位置查找(转基因材料个体)
- e. 研究SV在群体遗传进化中的作用(自然资源群体)
- f. 研究SV与选择驯化的关系(选择驯化群体与野生群体)
- g. SV GWAS(自然资源群体)
- 2.8 常规样品要求
- HMW(高分子质量的DNA):纯度、长度、总量
- 纯度要求
- 1) OD260/280 在 1.7-2.2 之间;
- 2) OD260/230 在 1.7-2.3 之间;
- 3) Nanodrop/Qubit 比值 0.8-2.5;
- 4) Nanodrop 检测峰图正常;
- 5) 澄清无色,无不可溶解物质;
- 6) 溶液中不含去污剂、 表面活性剂、 变性剂, 螯合剂和高浓度的盐;
- 7) 无单链 DNA、 RNA、 蛋白质或染料污染。
- 长度要求
- 1) 平均片段大小: PFGE 或低浓度琼脂糖(0.3%) 检测, >30kb;
- 2) 避免高速涡旋振荡等剧烈方式处理基因组 DNA, 如需混匀, 使用 宽口径枪头轻柔吹洗或颠倒轻叩样本管;
- 3) 切忌反复冻融
- 2.1 ONT测序平台介绍
- KASP基因分型
- 定义
- 竞争性等位基因特异性PCR分型系统(The Kompetitive Allele Specific PCR genotyping system)
- 目的
- 对SNP进行精准的双等位基因判断,从而进行大规模、快速的基因分型。
- 前提条件
- 1、有参物种(无参不建议接)
- 2、已知检测位点的SNP类型
- 3、2等位位点(只有2种碱基类型)
- 应用
- 可应用于QTL精细定位、分子辅助育种、种子资源鉴 定等工作,可以在短时间内准确判断分子标记类型。
- 技术原理
- 反应体系
- A、包含2条上游引物(识别不同的SNP),1条公共的下游引物; Allele 1引物:3'末端是C;5'末端与含FAM基团的引物序列相同; Allele 2引物:3'末端是A;5'末端与含HEX基团的引物序列相同;
- B、2条含荧光基团的特异引物和Taq polymerase; F探针与Allele 1标签序列一致,5'端有一个FAM荧光基团; H探针与Allele 2标签序列一致,5'端有一个HEX荧光基团; 相应于F探针和H探针,各设计一个3'端带淬灭基团的淬灭探针。
- 样品DNA:包含关注的SNP位点.
- 反应过程
- 第一轮PCR,模板会与可互补的(3'末端能配对的)特异的 上游引物进行退火,并发生扩增,这步完成SNP识别。同时 下游通用引物与模板结合并扩增。
- 第2轮PCR,下游通用引物扩增出带有Allele 1和Allele 2标签 序列的PCR产物,这步完成把特异标签序列引入与SNP对应 的PCR产物
- 第3轮PCR,以荧光基团的特异引物进行PCR扩增,一方面 淬灭探针被切碎,另一方面荧光探针更多地退火到新合成 的、没有淬灭基团的互补链上,发出荧光。通过检测荧光, 检出SNP位点上是哪个碱基
- 反应体系
- 适用群体
- 1、双亲杂交的遗传群体,F1、F2、F3、BC、RIL等,已知双亲的SNP分型,确定子代的未知基因型。
- 2、自然群体材料,因一次只能检测2种碱基,如为3或者4等位,还需再设计引物检测。
- 如何实现精细定位
- 主要结果
- 1、SNP位点鉴定报告书
- 2、每个SNP位点在每个样品的分型结果文件
- 3、每个SNP位点的引物信息及引物粉末
- 客户群
- 1、做基因定位的客户,多分布在农科院、农业大学、林业大学、水产学院等
- 2、B端客户,各种育种和种业公司,比如金色农华,大北农,奥瑞金,德农种业,屯玉种业等
- 送样要求
- 组织样
- 1、幼嫩叶片,大小(0.5cm×1cm)±0.2cm2;
- 2、叶片必须放入特定型号深孔96孔板,型号如下;
- 3、只接收叶片,不接收非叶片的植物组织样、动物、水产等组织样, 均需客户自提 DNA;
- 4、叶片按图示孔位摆放,客户记录好孔号和样品对应关系;
- 5、深孔板覆盖干净纸张,并用透明胶带封孔;
- 6、送散样需加收提取费。
- DNA
- 1、DNA提供量:20-50ng/μL浓度的DNA不少于200μL,具体用量将根据SNP位点数跟客户沟通;
- 2、KASP技术对DNA纯度要求不高,一般粗提的DNA即可,提取及溶解过程中尽量不要用到 EDTA, 如用到EDTA,建议将DNA稀释后使用,避免EDTA对KASP技术中酶活的影响。
- 3、最好把每个样品DNA转移至普通PCR板中,并用pcr板封口联排盖封盖好,避免漏样或混样;
- 4、如DNA放入普通离心管,需加收转样费。
- 组织样
- 报价
- 报价及计算公式
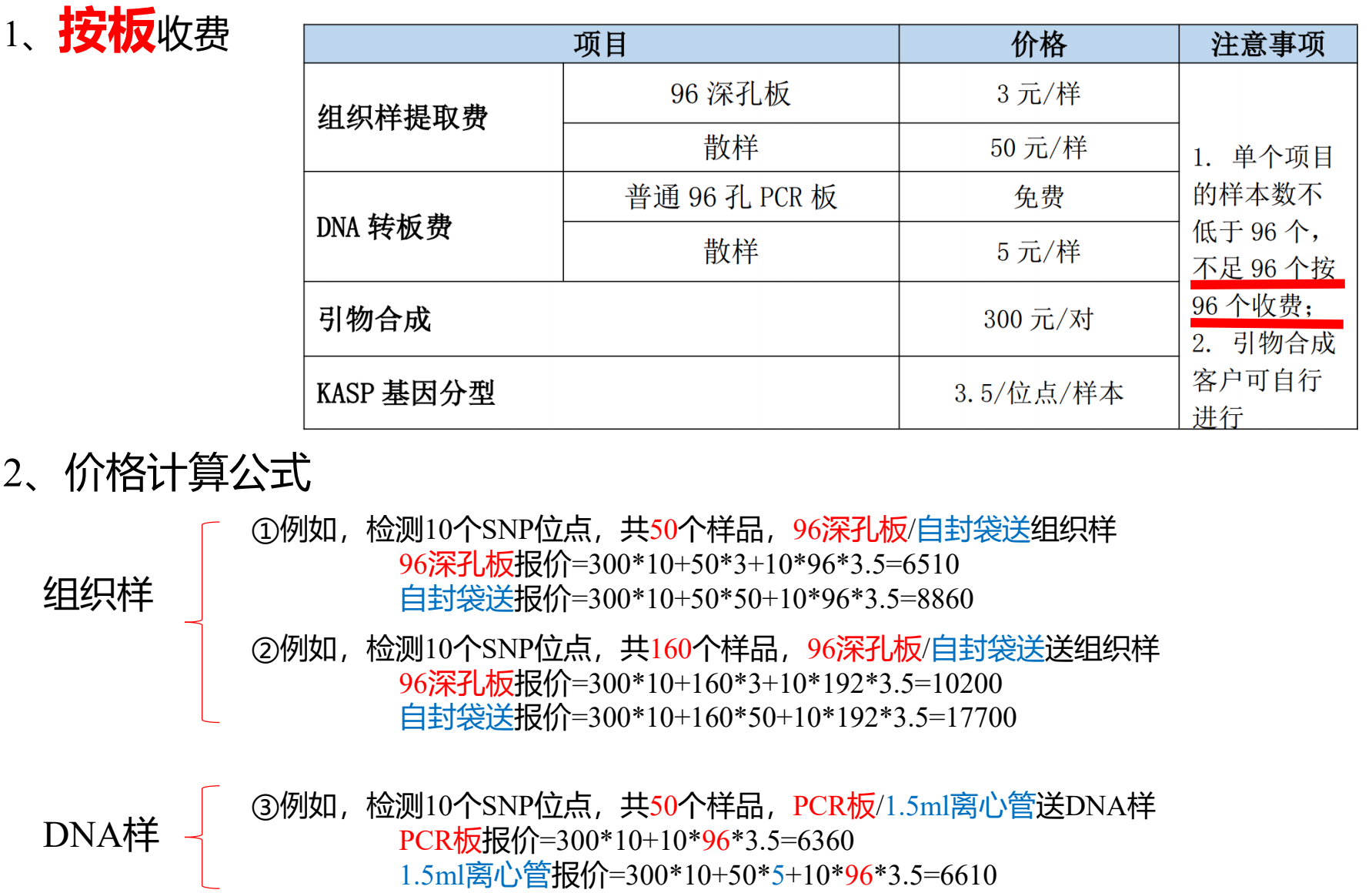
- 报价及计算公式
- 定义
- 个人总结
- 测序深度越深,假阳性越低
- 拷贝数变异属于结构变异(多次拷贝)
- 测序深度30X以上可以较为全面准确地获取复杂变异
- 遗传图谱中有大量相同片段,所以不需要高覆盖度
- 二代测SNP,三代重测序测SNP不准,主要做SV(大片段),如果都要做可以推基因组2+3


















 1087
1087
07-29

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











