










 本文全面概述了自然群体遗传进化分析与全基因组关联研究(GWAS)的方法,涵盖材料选择、常规与个性化分析手段,包括群体结构、主成分分析、系统进化树构建、选择性清除分析等,以及重测序、SLAF-seq和转录组测序在遗传进化和GWAS中的应用。
本文全面概述了自然群体遗传进化分析与全基因组关联研究(GWAS)的方法,涵盖材料选择、常规与个性化分析手段,包括群体结构、主成分分析、系统进化树构建、选择性清除分析等,以及重测序、SLAF-seq和转录组测序在遗传进化和GWAS中的应用。
- 自然群体研究方法概览
- 研究材料、方法、方向
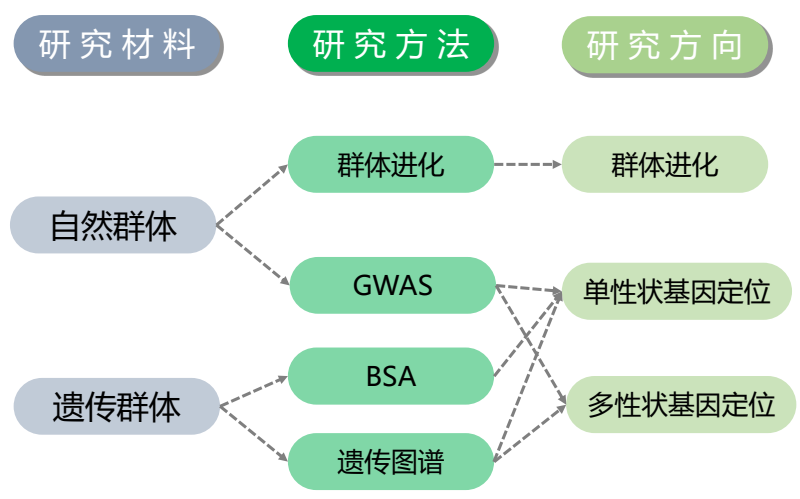
- 关系图
- 方法概览
- 遗传进化
- 进化\驯化历史 分化时间 地理起源
- GWAS
- 基因定位
- 遗传进化
- 关系图
- 研究材料、方法、方向
- 遗传进化分析
- 定义
- 通过系统发育分析揭示物种或种群间 的进化关系和发展历史。
- 研究目的
- 亲缘关系 动态分析(地理、分化、进化、历史) 表型性状
- 1.材料选择
- 选材来源
- ①不同进化阶段:祖先种→野生种→驯化种→改良种
- ②不同生境:不同地域、不同维度、不同海拔等
- 选材来源
- 2.常规分析
- 目的:
- 了解群体的真实构成情况
- 产生原因:
- 地理隔离——适应不同环境 、物种分化 、人工选择
- 方法:
- 群体结构分析
- 群体结构:指所研究的群体中存在基因频率不同的亚群。
- 方法:将群体分成K个服从Hardy-Weinberger平衡的亚群,将每个材料归到各个亚群,计 算每个材料其基因组变异源于第K个亚群的可能性
- K值为群体分群数,Admixture 软件一般以CV error最小值时K 为最佳分群;Structure软件以似然值最大时K为最佳分群。
- 不同颜色表示不同的分群
- 主成成份分析
- 主成分分析是一种数据降维的的分析方法,其基本思想是设法将原来众多的具有一定相关性的标 记重新组合成一组较少个数的互不相关的综合变量,这些变量即为主成分,从而最大程度的反映 原标记所代表的信息,保证新变量之间相互无关。一般会选择2个或者3个主成分对样品进行区分。
- 系统进化树
- 表明被认为具有共同祖先的各物种间进化关系树形图,是描述群体间分化顺序的分支图或树,用 来表示群体间的进化先后关系
- 建树方式:
- ①样品间SNP;
- ②样品间直系同源基因
- 建树方式:
- 无根树:描述不同样品间 的分类关系
- 有根树:体现不同样品间的分类关系以及进化先后关系
- 表明被认为具有共同祖先的各物种间进化关系树形图,是描述群体间分化顺序的分支图或树,用 来表示群体间的进化先后关系
- 选择性清除分析
- 当群体材料中发生自然变异,自然选择的作用将会促使有利变异在群体中被保留下来,其 两侧序列由于连锁效应同时被保留,而非有利变异则被选择清除,从而形成一段与自然选 择性状关联的基因组区间。
- 应用:
- 挖掘驯化过程中受选择的基因
- 挖掘物种适应性进化中受选择的基因
- 衡量指标:
- 群体内变异检验:π、 θ 、Hp
- 群体间变异检验:FST、XP-CLR、IHS、EHH等
- 中性选择:Tajima's D、Fu and Li's F
- 群体内变异检验
- π:核苷酸多态性
- 群体中任意两条不同个体的碱基差异数(即SNP)的平均值。多态性越低,受选 择程度越高。
- 性质:
- 1、一般情况下,群体π在0.001~0.01之间
- 2、受选择的群体,遗传多样性相对单一,π值较小;野生群体遗传多样性较大,π值较大
- π:核苷酸多态性
- 群体间变异检验
- FST(Fixation index):群体间遗传分化指数,通过比较群体间π值和亚群内π值的差异,来衡量种群 分化和遗传距离的一种方法。
- 性质:分化指数越大,差异越大。
- Fst居于0~1之间,越接近1表示两个群体之间分化程度越大,受选择程度越高,反之亦然
- 中性选择
- Tajima’s D:通过统计学模型来验证中性突变假说,理论值为零。
- 公式:Tajima’s D= (θπ–θW)/Var(θπ–θW) 越偏离0,受选择程度越高。
- >0,存在大量的中等频率的等位基因,推断群体瓶颈效应或者平衡选择。
- <0,存在大量的低频等位基因(稀有等位基因),推断定向选择或群体扩张。
- 连锁不平衡分析
- 群体内不同位点等位基因间的非随机性组合的关系,即当位于同一条染色体的两个等位基因(A,B)同 时存在的概率,大于群体中因随机分布而同时出现的概率时,就称这两个点处于LD状态。
- 作用
- 衡量
- 以r 2 值来度量连锁平衡状态
- 从0-1度量越高,LD越高,连锁不平衡越强
- 度量两个分析标记的基因型变化是否步调一致、存在相关性 的指标。
- 获得物种的最小的遗传单元(单体型)。
- 与驯化的关系
- 驯化程度越高,LD越大,LD衰减速度越慢。
- 群体结构分析
- 目的:
- 3.个性化研究
- 基因流分析
- 生物个体在迁移过程中致使基因从一个居群到另一个居群的过程,可发生在同种或不同种的生物种 群之间。
- 影响:增加种群内遗传变化、消弱种群间遗传差异
- 基因流分析
- 生物个体在迁移过程中致使基因从一个居群到另一个居群的过程,可发生在同种或不同种的生物种 群之间。
- 影响:增加种群内遗传变化、消弱种群间遗传差异
- 分化时间分析
- 以某一特定类群的化石记录作为参照点,通过基因序列间的分歧程度以及序列的平均置换速率 (分子钟)来估计速率恒定分支间的分歧时间,同时计算系统发育树上其它节点的发生时间, 从而推测相关类群的起源时间和不同类群的分歧时间。
- 种群历史动态
- 基于PSMC模型,阐明当前物种在历史进化过程中有效种群大小变化的过程,同时将历史上地质、 气候变化以及人类活动等历史事件发生时间段来解释有效群体大小剧烈变化的原因,有助于制定合 理有效的濒危物种保护策略。
- 基因流分析
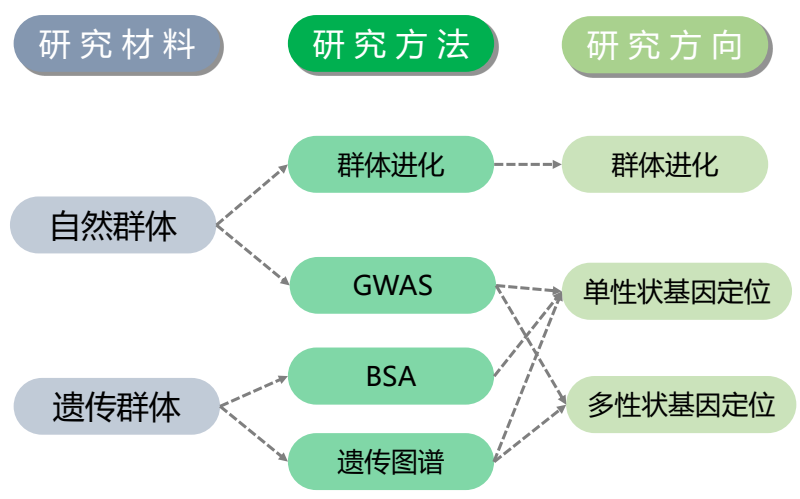
- 4.研究手段与方法
- 方法概览
- 方法图
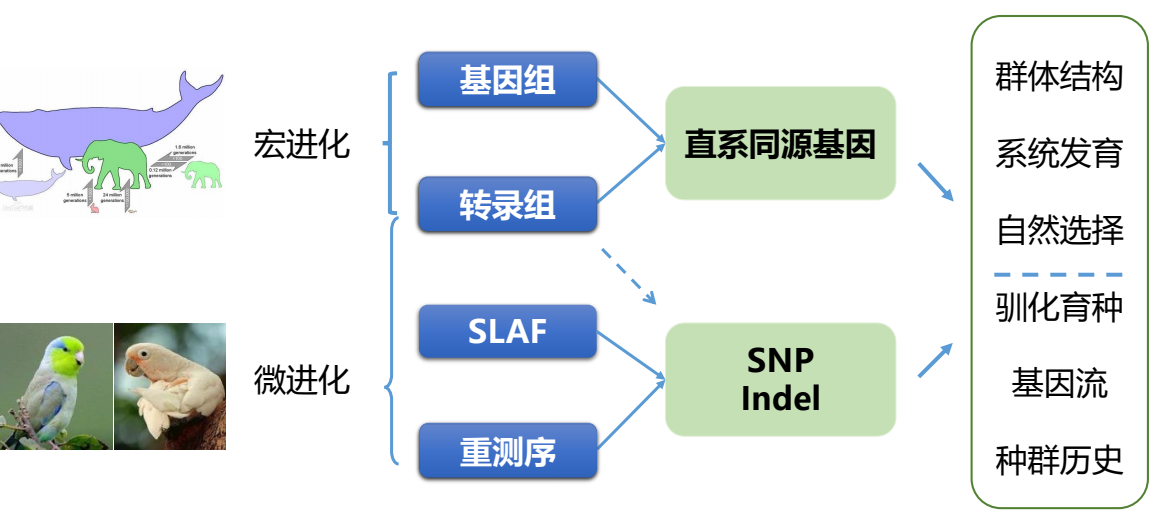
- 方法图
- 重测序遗传进化
- 适用情况:有参物种首推重测序
- 测序深度:推荐10x起
- 周期:3个月起
- 重测序-遗传进化
- 标准分析内容:
- 1、测序数据质控
- 2、与参考基因组比对统计 比对结果统计 插入片段分布统计 深度分布统计
- 3、全基因组变异检测 a)SNP位点检测及结果的注释 b)small InDel检测(≥10X)
- 4、 遗传多样性分析
- 5、群体分层分析
- a)利用NJ法构建系统发育树;
- b)群体结构分析(Admixture)
- c)PCA主成分分析
- 6、群体选择分析
- a)连锁不平衡分析
- b)选择清除分析(Fst,π,θ)
- c)受选择区域的基因功能注释
- d)受选择区域的基因的GO和KEGG富集
- 高级分析
- 1、利用其它软件进行系统发育树,群体结构分析;
- 2、基因流分析(TreeMix);
- 3、分子方差分析(AMOVA);
- 4、分化时间分析(Paml/beast2);
- 5、种群历史动态分析
- a)PSMC,要求分析的单个样品测序深度≥20×;
- b) smc++,要求群体内个体数不少于5个
- 标准分析内容:
- SLAF-遗传进化
- 适用情况:大基因组物种
- 测序指标

- 项目周期:75天起
- 预实验
- (1)电子酶切选择3种符合合同标签数的酶切组合
- (2)选择3个样品进行实际建库、测序、分析,每个样品3种酶切方案都做,相当于9个样品的试验
- (3)根据上述结果选择1种最优和最常用酶的组合作为正式实验的酶切方案
- (4)预实验周期45天
- 标准分析内容:
- (1)群体结构分析:PCA、群体分层、进化树
- (2)群体选择分析(有参)
- (2)自然选择位点筛选(无参)
- a) BayeScan、bayenv2、LFMM等软件进行全基因组范围内 自然选择位点筛选
- b) 多态标签或转录本Blast比对、GO等数据库注释筛选和环境 相关的基因,研究物种的适应性进化研究
- a)连锁不平衡分析
- b)选择清除分析(Fst,π,θ)
- c)受选择区域的基因功能注释
- d)受选择区域的基因的GO和KEGG富集
- 高级分析内容:
- (1)利用其它软件进行系统发育树,群体结构分析
- (2)遗传多样性分析(如PIC,He,Nei’s,Shannon 等参数)
- (3)基因流分析(TreeMix)
- (4)分子方差分析(AMOVA)
- (5)分化时间分析(Paml/beast2)
- 转录组-遗传进化
- 适用情况:有/无参物种,尤其适合无参物种
- 测序指标
-
- 项目周期:4个月起
- 标准分析内容:
- 有参
- (1) 基因功能注释
- (2) RNA-Seq整体质量评估
- (3) SNP和Indel分析
- (4) 群体分层分析
- a)NJ法构建系统发育树
- b)群体结构分析(Adimixture)
- c)PCA分析
- (5) 群体选择分析
- a)选择清除分析
- b)受选择区域的基因功能注释和富集
- c)受选择区域的基因的GO和KEGG富
- (6)基因表达量计算
- 无参
- (1)测序数据质控
- (2)转录组测序数据组装
- (3)Unigene功能注释
- (4)CDS预测
- (5)SNP检测
- (6)遗传多样性分析(如PIC,He,Nei’s,Shannon等参数)
- (7)群体分层分析
- a)利用NJ法构建系统发育树
- b)群体结构分析(Adimixture);
- c)PCA主成分分析;
- (8)自然选择位点筛选
- a) BayeScan、bayenv2、LFMM等软件进行全基因组范围内自然选择位点筛选
- b)多态标签或转录本Blast比对、GO等数据库注释
- c)筛选和环境相关的基因,研究物种的适应性进化研究基因表达量计算
- 有参
- 方法概览
- 定义
- GWAS研究
- 整体研究思路
- GWAS分析方法原理
- 在全基因组范围内选择遗传变异进行基因分型,统计分析每个变异与目标性状之间的关联性大小, 最终确认其与目标性状之间的相关性。
- 关联分析VS连锁分析
- (1)连锁分析需要构建F2、BC、RIL等群体,关联分析可用自然群体,大大缩短研究 年限
- (2)连锁分析每次只能分析同一位点的2个等位基因,联分析可以同时分析同一位点 的多个等位基因
- (3)连锁分析所用的群体小,在群体内发生的重组次数少,导致连锁作图的精度低, 而关联分析利用自然累计的重组,重组时间多,定位精度较高
- 技术路线图
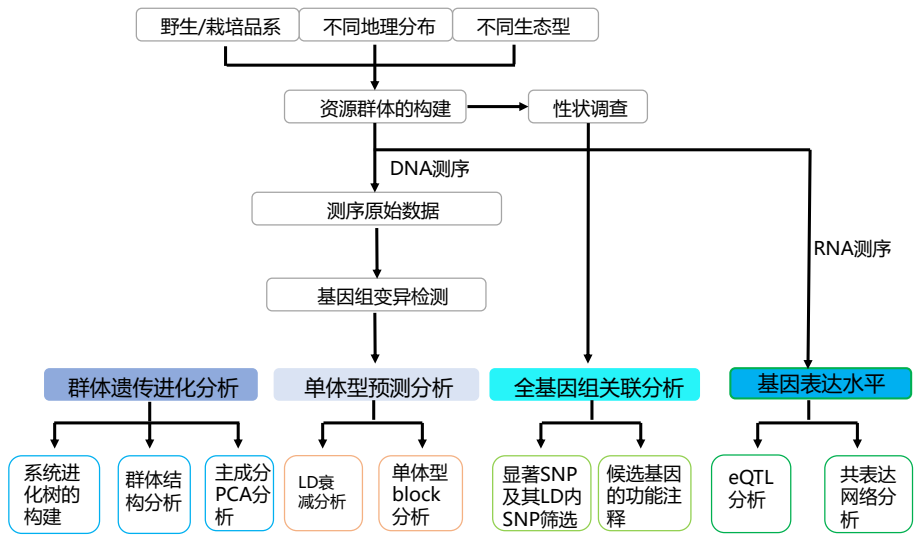
- 分析流程
- 材料选择
- 群体材料一个性状需要的样品数推荐大于200个
- 群体内的基因频率差异较小(群体没有分层)
- 性状表型变化明显
- 染色体倍性相同
- 推荐选材类型为:
- −种质资源库、自然群体、农家种 • 遗传变异丰富 • 注意避免个别种质遗传背景差异较大
- −家系群体(家系或多个家系混合) • 整体表型近似正态分布,多个家系混合时需要注意家系亲本间差异不能过大 • 最好构建可以专门用于GWAS遗传群体(如NAM群体和MAGIC群体)
- 性状调查
- 精确的表型检测是关联分析的关键
- GWAS针对数量性状和质量性状都适用 •
- 数量性状:多基因控制,能够测量得到具体数值,符合正态分布; 考虑到数量性状受环境影响大,建议将所有材料在同一环境下培 育或养殖,或者用多年多点的数据取平均或BLUP值作为性状值, 或用多个子代的数据取平均作为性状值。
- 质量性状:单基因控制,无法用具体数值衡量,可转换成0、1 等表示,需注意每个群体选取的样本量尽量相近。
- 基因分型
- 分型方式
- 重测序
- SLAF-seq
- RNA-seq
- 基因型填充
- 根据高深度测序样本的测序结果,去推导低深度样品缺失的基因型,从而提高所有样品的标记完整度基因型填充(针对小于3X的低深度重测序)。
- 分型方式
- 群体结构
- 5类群体结构
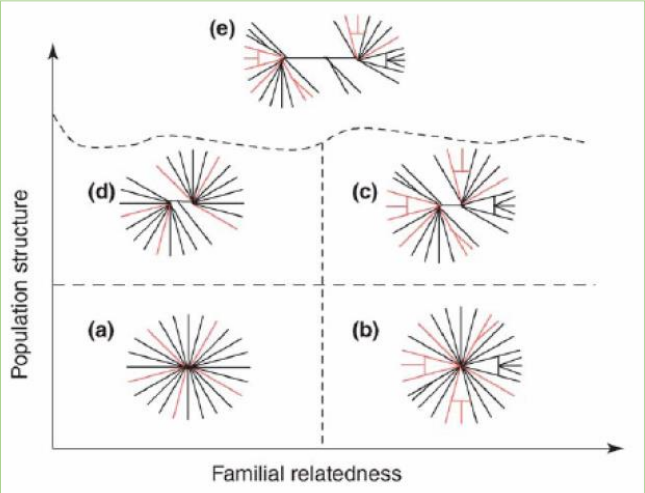
- 群体结构评估方法(之前有提及)
- 群体结构分析:帮助了解所研究的样 品属于几个亚群。 PCA分析:分析群体的进化关系,同 时可以检测离群样品。 系统发育分析:样品间的进化关系
- 5类群体结构
- 关联分析
- 分析模型
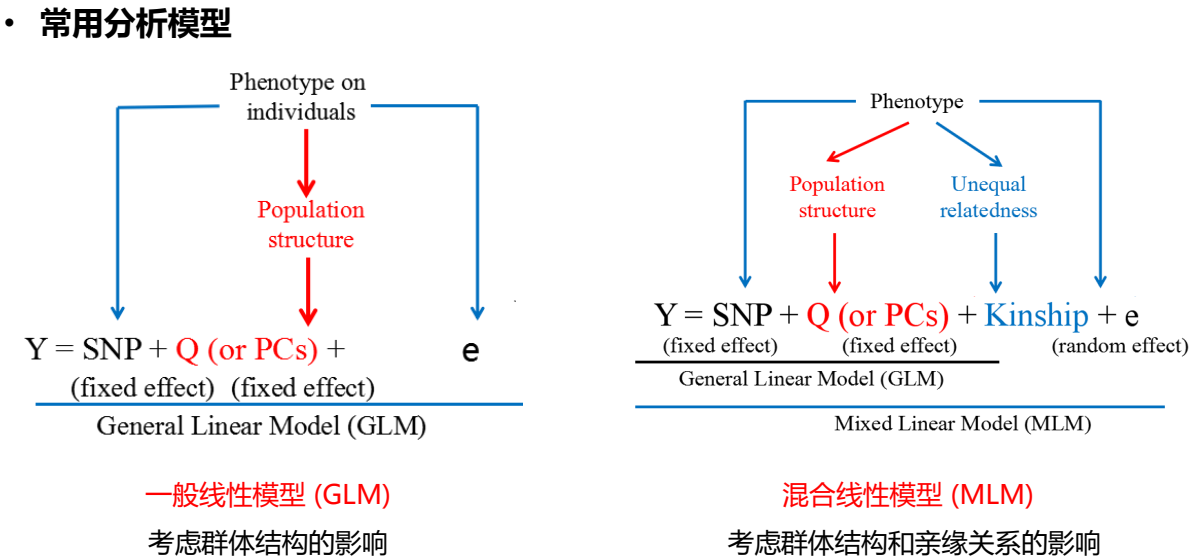
- 根据每个样品的表型值和分型的SNP信息,计算全基因组范围内所有SNP位点的 关联值(P值),P值超出阈值线的染色体区间即为定位结果。
- 分析模型
- 关联后研究内容
- 1)对显著位点附近进行LD Block分析,确定候选区间的范围
- 2)对候选区间内的基因进功能注释(包括NR,GO,KEGG等)
- 3)显著位点是否位于编码区,是否引起编码氨基酸的改变
- 4)同源分析,结合其他物种对应的同源基因的功能猜测候选基因的功能
- 材料选择
- 研究方法与手段
- 重测序-GWAS
- 适用情况:有参物种首推重测序
- 测序深度:推荐10x起
- 周期:3个月起
- 标准分析内容:
- 1、测序数据质控 2、与参考基因组比对统计 比对结果统计 插入片段分布统计 测序深度及覆盖度统计 3、全基因组变异检测 全基因组范围内的SNP和InDel检测 4、遗传进化分析 系统发育树构建 群体结构分析 主成分PCA分析 亲缘关系分析 5、LD连锁不平衡分析 6、全基因组关联分析 a)SNP与性状关联分析(GLM、MLM、CMLM、EMMAX、 FASTLMM模型 ) b)候选基因功能注释 7、基于候选位点的Block分析
- SLAF-GWAS
- 适用情况:有参大基因组物种
- 测序指标

- 项目周期:75天起
- 预实验
- (1)电子酶切选择3种符合合同标签数的酶切组合 (2)选择3个样品进行实际建库、测序、分析,每个样品3种酶切方案都做,相当于9个样品的试验 (3)根据上述结果选择1种最优和最常用酶的组合作为正式实验的酶切方案 (4)预实验周期45天
- 标准分析内容:
- (1)测序数据质控 原始数据统计 碱基测序质量分布 碱基类型分布 低质量数据过滤 数据统计与数据质量评估 (2)SNP检测与注释 样品之间SNP的检测 SNP结果注释(有参) (3)遗传进化分析 构建系统发育树 群体结构分析 主成分分析 亲缘关系分析 (4)LD连锁不平衡分析 (5)全基因组关联分析及基因注释(有参) a)SNP与性状关联分析(GLM、MLM、CMLM、EMMAX、 FASTLMM模型 ) b)候选区间的基因注释 (6)基于候选位点的Block分析(有参)
- 转录组-GWAS
- 适用情况:有参物种,尤其适合大基因组物种
- 测序指标

- 项目周期:3个月起
- 标准分析内容:
- 1、测序数据质控与统计 2、与参考基因组比对分析 3、SNP的检测 4、基因表达量计算 8、遗传结构分析 a)构建系统发育树 b)群体结构分析 c)主成分分析 d)亲缘关系分析 9、关联分析 a)SNP关联分析(GLM、MLM、CMLM、EMMAX、FASTLMM模型 ) b)候选基因功能注释 10、候选基因的功能富集分析
- 高级分析内容:
- 1、eQTL分析 a)样品间差异基因检测 b)eQTL定位 2、基因共表达网络分析
- 重测序-GWAS
- GWAS分析方法原理
- 整体研究思路
- 百迈客经验介绍
- 经验丰富
- 发表文章多
- 个人总结
- BSA是分离群体分组分析法
- 高级分析就是个性化分析
- 如果老师打算做SNP研究,可以尽量推荐遗传进化,把单子做;或者反过来,如来想做遗传进化,可以用转录组或者SLAF组合出单
- 无论用哪种方式进行基因分型,最终都是要得到SNP位点
- 欢迎进入生信流程群讨论:




















 3340
3340

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











