









非负矩阵分解(NMF)和一致性聚类(ConsensusClusterPlus)是两种常用的聚类和模式识别方法,它们在算法原理、使用场景和结果解读上都有相似和不同之处。这两种代码流程已经被诸多老师所解析和展示,本次也是跟着老师们发布在网上的流程进行练习和整理。
非负矩阵分解和一致性聚类的异同点
非负矩阵分解(NMF)
使用场景:
NMF主要用于从高维数据中提取潜在模式或特征,例如基因表达数据中的特征模块识别,或者文本数据中的主题提取。
算法原理:
NMF是一种矩阵分解技术,将一个非负矩阵 VVV 分解为两个非负矩阵 WWW 和 HHH,即 V≈W×HV \approx W \times HV≈W×H。其中 WWW 表示特征与重要因子的关系, HHH 表示重要因子与样本的关系。通过这种方式,NMF可以将复杂的高维数据表示为较低维度的潜在结构。
结果解读:
分解后的矩阵 WWW 和 HHH 可以用于理解特征和样本之间的关系,从而识别数据中的潜在模式。通常通过热图或聚类分析对分解结果进行可视化。
一致性聚类(ConsensusClusterPlus)
使用场景:
ConsensusClusterPlus是用于评估聚类稳定性和确定最佳聚类数目的工具,特别适合用于样本聚类分析,如基因表达数据中的样本分类。
算法原理:
ConsensusClusterPlus通过多次重复聚类(例如K-means、层次聚类等)并计算样本之间的一致性矩阵来评估聚类结果的稳定性。通过这种一致性评估,可以确定聚类结果的可靠性和最适合的数据分群数目。
结果解读:
该方法的结果通常包括一致性矩阵、一致性得分曲线、聚类数目评估图等。这些结果帮助用户确定最佳聚类数目,并评估聚类的稳定性和准确性。
异同点
相似点:
● 都可以应用于生物数据分析中,例如基因表达数据的模式识别和样本聚类。
● 两者都需要用户输入数据矩阵并通过一定的算法处理来得到聚类或模式。
不同点:
● 目的:NMF侧重于特征或模式提取,而ConsensusClusterPlus侧重于评估和验证聚类结果的稳定性和最佳聚类数目。
● 算法:NMF基于矩阵分解,而ConsensusClusterPlus基于多次聚类和一致性评估。
● 输出:NMF输出特征与样本的关联矩阵,而ConsensusClusterPlus输出一致性矩阵及相关评估图表。
● 使用方式:NMF一般用作数据预处理的一部分,提取特征后再进行聚类分析,而ConsensusClusterPlus本身就是一种聚类验证工具,直接用于确定最佳聚类方案。
总结来说,NMF和ConsensusClusterPlus在数据分析中具有不同的用途和方法,但它们都可以用于揭示数据中的潜在结构或模式。NMF侧重于提取数据中的重要因子/特征,而ConsensusClusterPlus则侧重于验证聚类的稳定性和一致性。
NMF实操
在实操前还需要稍微了解一下NMF中提供的算法
1. Brunet 算法
描述: 基于 Kullback-Leibler 散度的标准 NMF 算法,使用乘法更新规则。
适用场景: 这种算法特别适用于聚类分析,尤其是在生物信息学中用于基因表达数据的亚型识别。Brunet 算法通过多次运行 NMF,并生成共识矩阵来稳定结果,使其在生物学数据的聚类稳定性方面表现良好。如果你正在分析基因表达数据,并希望得到稳健的聚类结果,Brunet 算法是一个好的选择。
2. Lee 算法
描述: 基于欧几里得距离的标准 NMF 算法,也使用乘法更新规则。
适用场景: Lee 算法适用于大多数标准的 NMF 分解任务,特别是在数据没有明显的稀疏性需求时。它是最基础和广泛使用的 NMF 算法,适合用于探索数据的潜在结构或主题分解。如果你需要对任何数据集进行一般的矩阵分解,且不需要考虑稀疏性或稳定性问题,可以选择 Lee 算法。
3. nsNMF (Non-smooth NMF)
描述: 使用修改版的 Lee 和 Seung 的乘法更新规则,旨在生成更稀疏的矩阵。 适用场景: 当你的数据具有稀疏性特征,或者你希望在分解结果中保留更多的稀疏性时,nsNMF 是一个不错的选择。这种算法通过引入非平滑性(non-smoothness)约束,使得结果矩阵中包含更多的零元素,适用于稀疏数据或当你希望分解结果中的基因特征矩阵更具可解释性时。
4. Offset NMF
描述: 使用修改版的 Lee 和 Seung 的乘法更新规则,适合包含截距项的 NMF 模型。 适用场景: 当你处理的数据需要在模型中引入截距项来调整基准水平时,可以考虑使用 Offset NMF。这个算法主要用于需要调整数据基线的场景,例如在某些基因表达分析中,调整表达值的基线可能会更好地反映生物学意义。
5. PE-NMF (Pattern-Expression NMF)
描述: 一种针对模式表达的 NMF 算法,使用乘法更新来最小化基于欧几里得距离的目标函数,并对模式表达进行正则化。
适用场景: PE-NMF 特别适用于需要识别数据中的模式或表达特征的场景。它通过对模式表达进行正则化,可以提高模式的解释性和稳定性,特别适用于基因表达数据分析,帮助识别不同的表达模式。如果你的分析目标是发现数据中的特定模式或表达结构,PE-NMF 是一个值得考虑的选项。
6. SNMF (Sparse NMF) / ALS (Alternating Least Squares)
描述: 使用交替最小二乘法(ALS)进行 NMF 分解,速度非常快,适合处理大规模数据。
适用场景: SNMF/ALS 适用于处理大规模数据集,尤其是在需要快速分解和处理数据时。由于 ALS 方法在每一步中都可以处理大块数据,计算效率高,适合在大数据分析或实时处理场景中使用。如果你有一个大规模的数据集,并且对计算效率有较高要求,可以选择 SNMF/ALS 方法。
选择合适的 NMF 算法需要根据具体分析需求、数据特点以及对结果的期望来决定。一般来说,最常用的还是Brunet和Lee算法。
分析流程
1、导入数据
rm(list = ls())
proj = "GSEXXX"
dir.create("./1-GSEXXX-NMF")
setwd("./1-GSEXXX-NMF")
load(paste0(proj,".Rdata"))
# check一下
head(exp)[1:4,1:4]
BI_002 BI_004 BI_005 BI_006
A1BG 4.375762 3.690328 3.384776 3.453303
A1BG-AS1 3.118460 1.761581 2.718007 2.695267
A2M 8.851277 10.339066 11.085478 9.540225
A2M-AS1 5.780874 7.786958 6.180915 5.616252
exp <- log2(edgeR::cpm(exp)+1)
#提取PH数据
exp <- exp[,Group == "tumor"]
2、确定NMF分组
# 进行非负矩阵分解,评估rank值的选取
library(NMF)
ranks <- 2:10
range(exp)
exp <- exp[rowSums(exp) != 0, ] # 移除全为零的行
system.time({
result <- nmf(exp,ranks,nrun=50)
})
# user system elapsed
# 5678.5 5165.05 10843.55
save(result,file = "NMF.Rdata")
load("NMF.Rdata")
plot(result)
多个rank且nrun设置的比较大的时候运行起来真的非常非常慢。10843.55秒...
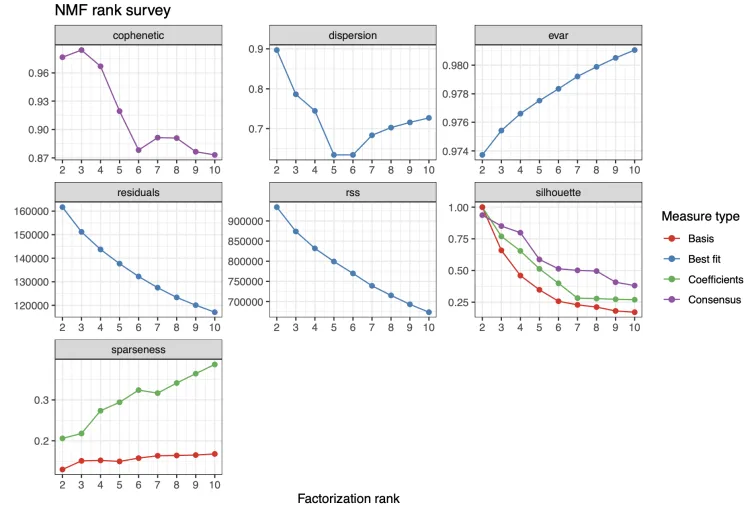
在非负矩阵分解(NMF)中,rank值通常代表因子分解的维度或因子的数量。选择合适的rank值对于确保模型的有效性和稳定性至关重要。
主要性能指标解释
1. 残差平方和(RSS, Residual Sum of Squares):
● 定义: RSS 是数据与模型拟合结果之间误差的平方和。较小的 RSS 表明模型更好地拟合了数据。
● 选择rank: 随着 rank值的增加,RSS 通常会减少。然而,随着rank的不断增加,RSS 的减少速度会变慢,表现为一个“肘部”(elbow)点。通常,在这个“肘部”点附近选择 rank 值,这是一个平衡模型复杂性和拟合效果的最佳点。
2. 一致性系数(Cophenetic Correlation Coefficient):
● 定义: 一致性系数用于衡量聚类结果的一致性和稳定性。值越高,聚类结果越稳定。
● 选择rank:通常选择一致性系数较高时的rank值,因为这表明在该 rank下模型具有较高的稳定性。
3. 轮廓系数(Silhouette Score):
● 定义: 轮廓系数是衡量聚类质量的指标。值范围在 -1 到 1 之间,越高表示聚类质量越好。
● 选择rank:一般选择轮廓系数最高时的rank值,这表明在该 rank下模型的聚类效果最佳。
4. 分散性(Dispersion):
● 定义: 分散性表示聚类的紧密程度。分散性越小,表示聚类结果越紧密。
● 选择rank:选择分散性最低的rank 值,通常能确保模型在该 rank下的聚类紧密性最好。
5. 稀疏性(Sparseness):
● 定义: 稀疏性衡量矩阵的稀疏程度。稀疏性越高,矩阵中的大多数元素接近零,这可以提高结果的可解释性。
● 选择rank:如果解释性是模型的主要目标,可以选择稀疏性较高时的 rank值。
如何选择合适的rank
观察指标的趋势:
● 在选择rank 时,可以绘制每个指标随rank变化的曲线,并观察其趋势。
● 找到“肘部”点,一致性系数最高点,轮廓系数最高点,分散性最低点,以及稀疏性高点。
综合考虑:
● 通常情况下,选择一个能够平衡多个指标的rank值。例如,一个在 RSS 曲线肘部且一致性系数和轮廓系数较高的 rank值可能是最佳选择。
模型验证:
● 选择多个候选 rank 值,通过交叉验证或其他模型验证方法评估模型在这些 rank 下的性能,最终确定最优的rank。
最常用标准:cophenetic 曲线下降范围最大的前点。
4、NMF分析-确定最佳rank之后再进行分析
# rank=4,进行NMF分析
seed = 24812
res_rank4 <- nmf(exp,
rank = 4,
nrun= 10,
seed = seed,
method = "lee")
## 提取特征基因
index <- extractFeatures(res_rank4,"max") # 能给出选了哪些关键的基因,可以用这些基因再次聚类
exp_rank4 = exp[unlist(index),]#提取特征基因
exp_rank4 = na.omit(exp_rank4)
nmf_rank4 <- nmf(exp_rank4,
rank = 4,
seed = seed,
method = "brunet")
group <- predict(nmf_rank4) # 提出亚型
table(group)
#设置颜色
jco <- c("#2874C5","#EABF00","#C6524A","#868686") #, "#FF0000FF","#CCFF00FF")
sig.order <- unlist(index)
NMF.Exp.rank4 <- exp[sig.order,]
NMF.Exp.rank4 <- na.omit(NMF.Exp.rank4) #sig.order有时候会有缺失值
group <- predict(nmf_rank4) # 提出亚型
table(group)
png("consensusmap1.png", width = 1200, height = 1200,res = 300)
consensusmap(nmf_rank4,
labRow = NA,
labCol = NA,
annCol = data.frame("cluster"=group[colnames(NMF.Exp.rank4)]),
annColors = list(cluster=c("1"=jco[1],"2"=jco[2],"3"=jco[3],"4"=jco[4])))#,"5"=jco[5],"6"=jco[6])))
dev.off()
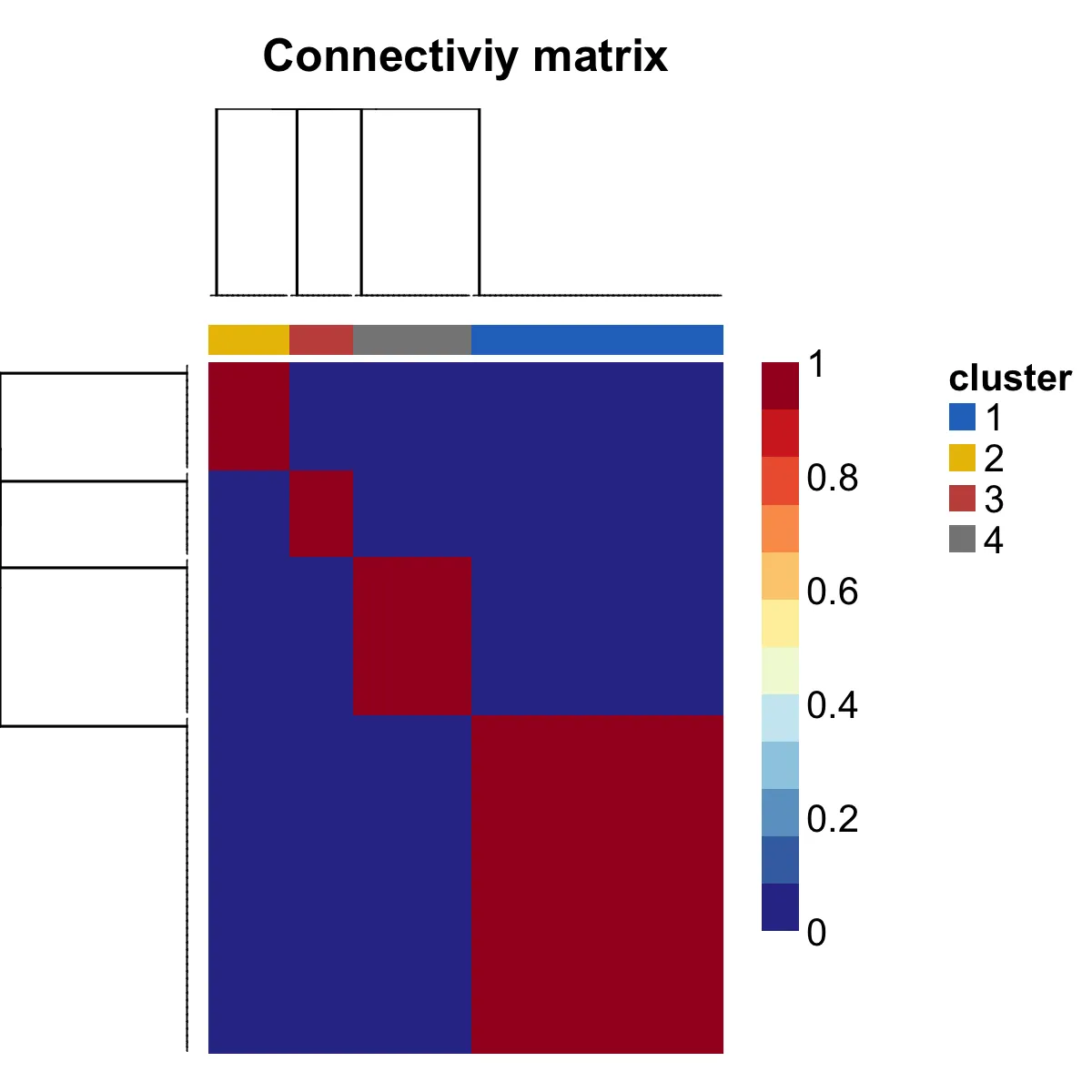
这里是对NMF分析进行了再次聚类:
这是由于初始NMF分析可能包括了一些噪声或不太相关的基因,再次对提取出的特征基因进行NMF分析有助于精炼聚类结果。这种做法可以帮助你进一步细化聚类,排除噪声,增强模型的鲁棒性。同时再次NMF分析后,可以通过系数矩阵中的模式识别潜在的亚型。这些亚型可能比第一次分析中获得的更为准确和一致。
在实际操作过程中,笔者遇到过一个亚型只有一个样本的情况,这时候通过再次聚类就可以合并这个样本,或者也可以在初始分析的时候去除这个样本。
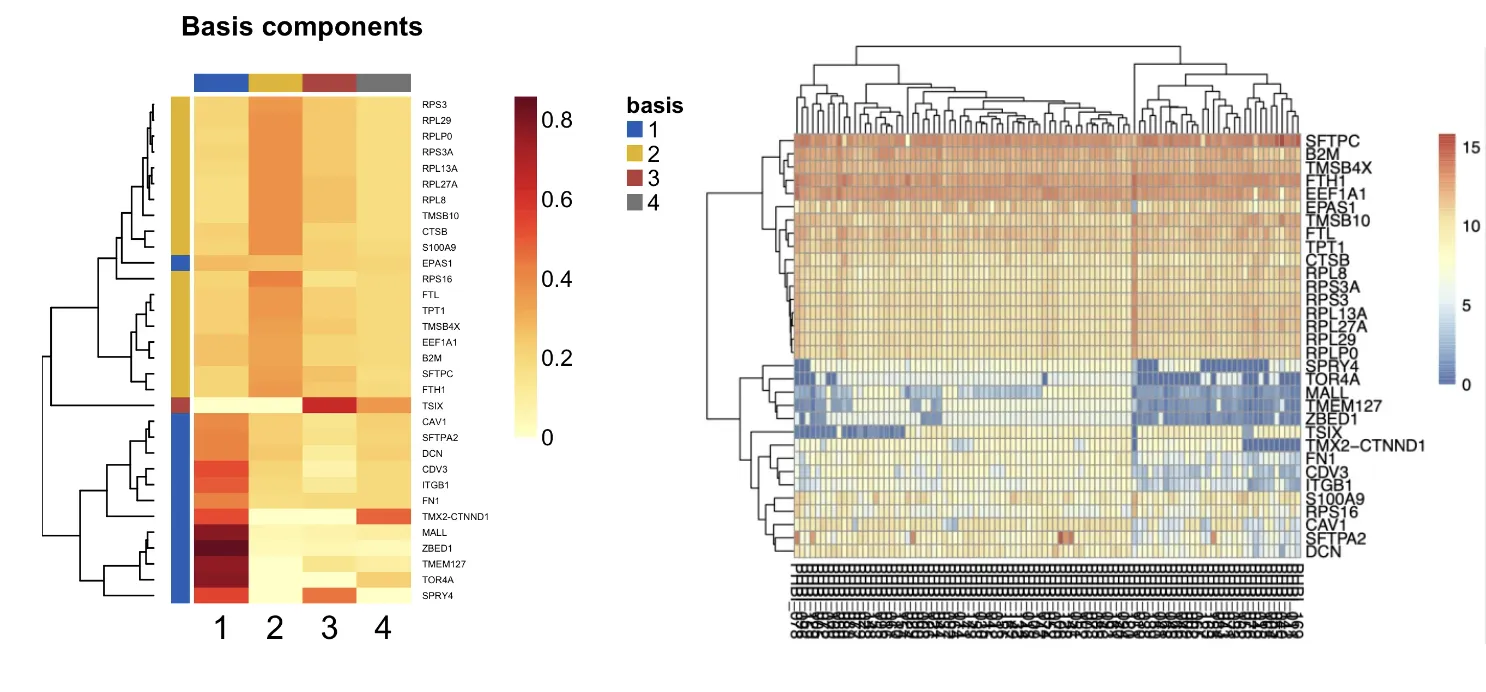
5、其他热图的绘制
## 01从这张图可以比较清晰地看到各亚型中的驱动signature(深色),与下面的nmf heatmap对应
png("consensusmap.png", width = 1200, height = 1200,res = 300)
basismap(nmf_rank4,
cexCol = 1.5,
cexRow = 1.5,
annColors=list(c("1"=jco[1],"2"=jco[2],"3"=jco[3],"4"=jco[4])))
#,"5"=jco[5],"6"=jco[6])))
dev.off()
## 02绘制常规热图
library(pheatmap)
pheatmap(exp_rank4)
## 02按照样本聚类顺序提供样本名
head(exp_rank4[1:6,1:6]) #刚开始是按照连续顺序的
sample.order <- names(group[order(group)])
exp_rank4=exp_rank4[,sample.order]
head(exp_rank4[1:6,1:6]) ##调整成样本聚类顺序
## 03 添加注释信息,绘制热图
ac=data.frame(group=group)
ac$samples=rownames(ac)
ac1=ac[order(ac$group),]
identical(colnames(exp_rank4),rownames(ac1))
## [1] TRUE
pheatmap(exp_rank4,show_colnames =F,show_rownames = T,annotation_col=ac1,cluster_cols = FALSE)
ConsensusClusterPlus实操
在实操前还需要稍微了解一下ConsensusClusterPlus中提供的算法
1. K-means 聚类 (kmeans)
描述: 一种经典的聚类算法,它将数据分成 K 个簇,目的是最小化簇内方差。K-means 使用迭代优化的方式,将数据点分配到最接近的簇中心。
适用场景: 适用于中小型数据集,特别是在簇的形状比较规则(如球形)时表现较好。广泛应用于各类数据集的初步聚类分析。
2. 层次聚类 (Hierarchical Clustering)
描述: 层次聚类逐步将数据点合并成簇(凝聚型)或分裂成更小的簇(分裂型),直到满足停止条件为止。可以生成一个聚类树(树状图),显示不同聚类的嵌套关系。
适用场景: 常用于需要层次结构的聚类分析,如基因表达数据的分析。适用于希望理解数据点之间的层次关系或自然嵌套结构的场景。
3. PAM 聚类 (Partitioning Around Medoids)
描述: PAM 是基于 K-medoids 的聚类方法,类似于 K-means,但使用数据点本身作为聚类中心(medoids)。PAM 对噪声和离群值有更好的鲁棒性。
适用场景: 适用于存在噪声和离群点的数据集,比 K-means 更稳健,适合处理包含异常值的数据。
4. 基于模型的聚类 (Model-based Clustering)
描述: 假设数据来自于多个不同分布的混合体,使用概率模型(如高斯混合模型)来拟合数据,并根据最大似然估计分配数据点到不同的簇。
适用场景: 适用于希望通过假设数据生成机制来进行聚类的场景,尤其是数据可能具有复杂分布结构时。
5. 谱聚类 (Spectral Clustering)
描述: 谱聚类基于图论,通过将数据点映射到低维空间后进行 K-means 聚类来识别簇。它通过计算数据点之间的相似度图谱来进行聚类。
适用场景: 适用于处理非线性结构的复杂数据集,尤其是高维数据或存在明显非线性分离的数据集。
6. 自组织映射 (Self-organizing map, SOM)
描述: 基于神经网络的一种无监督学习方法,用于将高维数据映射到低维空间。SOM 通过训练神经元的权重来发现数据的内在结构,并将相似的数据点聚集在一起。
适用场景: 适用于需要数据可视化和降维的场景,如基因组数据分析。特别适合高维数据且希望同时进行聚类和可视化的场景。
在使用ConsensusClusterPlus时,可以根据数据的特性和分析目标选择合适的聚类算法。例如,如果数据具有复杂的非线性结构,可以考虑谱聚类;如果需要处理噪声或离群点,PAM 是一个较好的选择;如果希望在不指定簇数的情况下进行分析,可以选择层次聚类。K-means最常用。
分析流程
1、导入数据-同样的数据
rm(list = ls())
proj = "GSEXXX"
dir.create("./1-GSEXXX-NMF")
setwd("./1-GSEXXX-NMF")
load(paste0(proj,".Rdata"))
# check一下
head(exp)[1:4,1:4]
exp <- log2(edgeR::cpm(exp)+1)
#提取PH数据
exp <- exp[,Group == "tumor"]
meta <- clinical[Group == "tumor",]
#提取mad值排前5000的基因
mads <- apply(dat,1,mad)
dat <- dat[rev(order(mads))[1:5000],]
par(mfrow = c(1,2))
boxplot(dat[,1:20],main = "before")
dat <- sweep(dat,1, apply(dat,1,median,na.rm=T))
boxplot(dat[,1:20],main = "after")
2、ConsensusClusterPlus
library(ConsensusClusterPlus)
maxK <- 9 #最多分成几组
results <- ConsensusClusterPlus(dat,
maxK = maxK,
reps = 500,
pItem = 0.8,
#tmyPal = c("white","#tomato"),
pFeature = 1,
clusterAlg = "km",
seed = 24812,
title="ConsensusCluster/",
distance="euclidean",
innerLinkage="complete",
plot="pdf")
icl = calcICL(results,
title="ConsensusCluster",
plot="pdf")
ConsensusClusterPlus参数介绍:
dat: 输入的数据矩阵。通常行代表样本,列代表特征或变量。是进行聚类分析的基础数据。
maxK: 该参数表示聚类分析时测试的最大簇数 (K)。通常设定一个合适的范围,比如2到10,以确定数据的最佳聚类数。
reps: 重复聚类的次数。默认值为 100。增加重复次数可以提高一致性评估的准确性。在这里,设为500表示每个K值的聚类过程将重复500次。
pItem: 每次子采样时抽取样本的比例。默认值为 0.8。用于创建子样本来评估聚类的稳定性。pItem = 0.8表示每次采样80%的样本。
pFeature: 每次子采样时抽取特征(变量)的比例。默认值为 1。用于随机选取特征来进行聚类分析。pFeature = 1表示每次使用全部的特征。
clusterAlg: 选择的聚类算法。此参数决定使用哪种算法进行聚类。常见的选项包括 "km"(K-means 聚类),"hc"(层次聚类)等。在这里,"km"表示使用 K-means 聚类算法。
seed: 设置随机种子。确保结果的可重复性。相同的种子值会产生相同的随机样本。
title: 输出文件的保存路径和文件名前缀。设置结果文件的存储路径,通常用于保存聚类分析的图表和结果文件。在这里,所有输出文件将保存到 ConsensusCluster/ 目录下。
distance: 计算样本之间距离的方式。决定用于聚类分析的距离度量标准。常见选项包括 "euclidean"(欧氏距离),"manhattan"(曼哈顿距离)等。在这里,"euclidean"表示使用欧氏距离。
innerLinkage: 聚类分析中使用的链接方法。用于层次聚类中指定链接方法。常见的选项包括 "complete"(完全链接),"average"(平均链接)等。在这里,"complete"表示使用完全链接。
plot: 输出图形的文件格式。指定聚类结果图形输出的格式。选项包括 "pdf"、"png"、"tiff"等。在这里,选择 "pdf" 表示输出 PDF 格式的图形文件。
如何选择合适的clusters
1. 共识矩阵 (Consensus Matrix) 和CDF曲线:
● 每个k值都有一个对应的共识矩阵。这些矩阵展示了在多次聚类中,样本是否一致地被分配到相同的簇中。
● 理想情况下,共识矩阵的图像应该在每个簇中显示出清晰的块状结构,表示簇内的样本高度一致(白色模块里边没有蓝色,蓝色模块里边没有白色)。
● 另外,共识CDF曲线可以帮助我们选择k值。通过比较不同k值下的CDF曲线变化,当CDF曲线的变化幅度开始趋于平缓时(即Δ面积变小),说明增加k值带来的聚类稳定性提高不明显,这时候的k值可能是合适的选择。

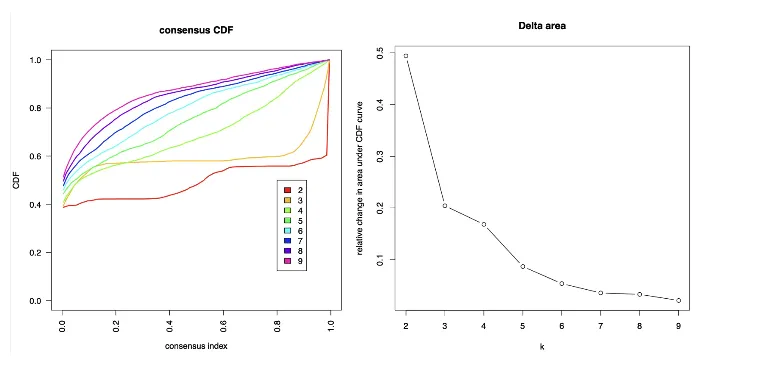
2. ICL (Item Consensus Level):
● 可以看到不同k值下的项目一致性水平(item consensus level)和簇一致性(cluster consensus)。
● ICL图帮助评估聚类质量。较高的簇一致性值表明聚类结果在重复中是稳定的。
● 在多个k值中,选择ICL数值较高且变化不大(即连续多个k值ICL变化较小)的一组作为最终的分群数目。
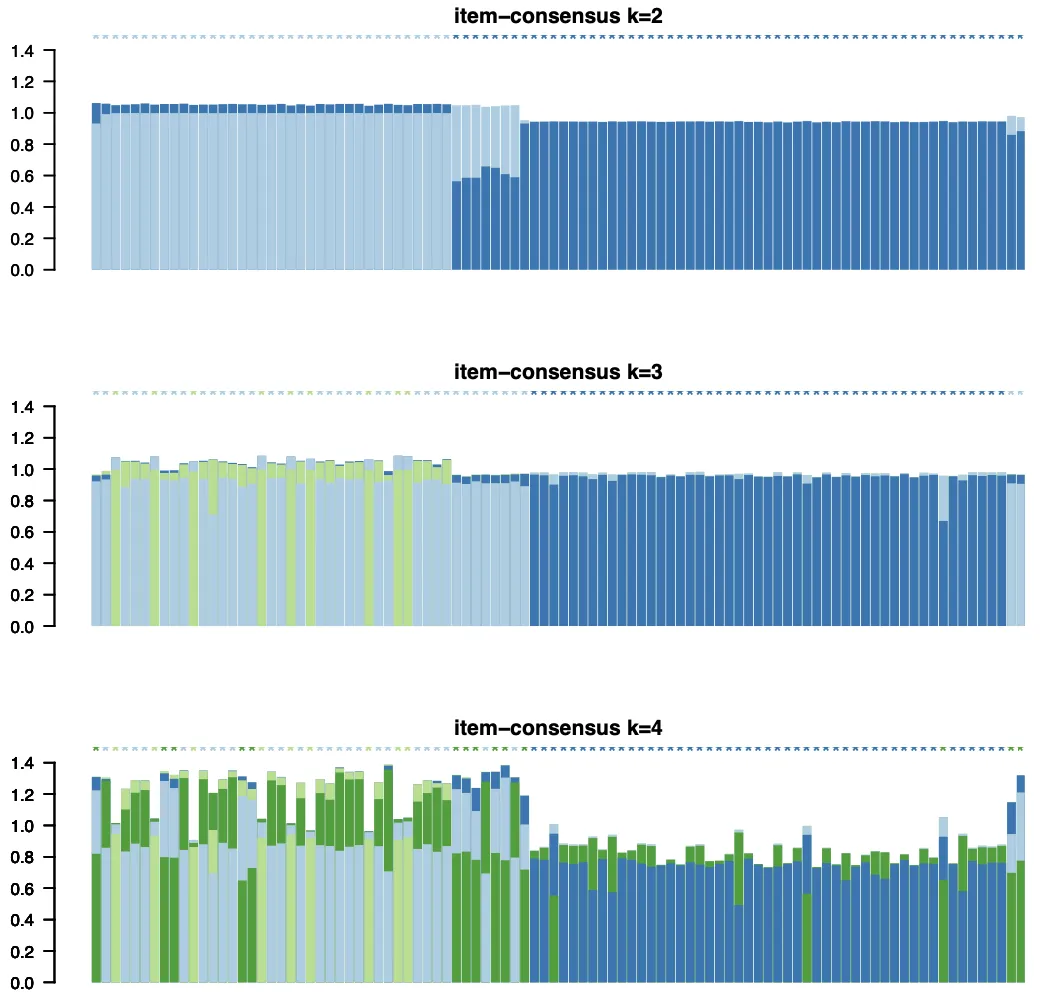

3、pca标准筛选
如果看图觉得很难分辨的话可以采用PCA的方法
Kvec = 2:maxK
x1 = 0.1; x2 = 0.9 # threshold defining the intermediate sub-interval
PAC = rep(NA,length(Kvec))
names(PAC) = paste("K=",Kvec,sep="") # from 2 to maxK
for(i in Kvec){
M = results[[i]]$consensusMatrix
Fn = ecdf(M[lower.tri(M)])
PAC[i-1] = Fn(x2) - Fn(x1)
}#end for i
# The optimal K
optK = Kvec[which.min(PAC)]
optK
这个可以辅助确认cluster的数量,也可以在难以分辨的时候帮助划分cluster
4、cluster提取
library(tidyverse)
table(results[[optK]]$consensusClass)
Cluster = results[[optK]]$consensusClass
identical(names(Cluster),rownames(meta))
meta$cluster <- Cluster
亚型提取完之后就可以来一套连招,把亚型贴到样本分组上去,然后进行生存分析~ 选择预后较差的亚群进行重点分析啦~
笔者对这两个方法的理解:
1、这两个方法从宽泛的角度来说都可以看作是亚群划分的方法;
2、非负矩阵分解能够更好的挑选出不同亚群的显著特征基因;
3、一致性聚类能够将将样本稳定的聚类分群,但是也许会掩盖里边的重要特征(可另一个角度来说重要特征一定具有泛化性吗,且极少的特征基因一定具有重大功能吗?);
4、独立使用时,两者同时使用并比较,选择其中一个方法对数据进行亚群划分;
5、联合使用时,应按照先后顺序进行使用,比如可采用非负矩阵分解先对样本进行分群,确定特征基因,锚定是何种亚群,之后再采用一致性聚类对亚群内部进行再次分群。有点像高考的时候先报不同的专业(非负矩阵分解),然后考研的时候在大专业的框架下选择细分专业(一致性聚类)。
参考资料:
1、NMF:https://nmf.r-forge.r-project.org/vignettes/NMF-vignette.pdf
2、ConsensusClusterPlus:https://bioconductor.org/packages/release/bioc/vignettes/ConsensusClusterPlus/inst/doc/ConsensusClusterPlus.pdf
3、生信菜鸟团: https://mp.weixin.qq.com/s/LQJp9Q7cZF2VxuXK1l8Pow
4、生信星球: https://mp.weixin.qq.com/s/L1UAnBzTqivlOPmMVH9pRQ
致谢:感谢曾老师,小洁老师以及生信技能树团队全体成员。
注:若对内容有疑惑或者有发现明确错误的朋友,请联系后台(欢迎交流)。更多内容可关注公众号:生信方舟
- END -

















 1万+
1万+

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









