







从样本准备到寄送公司,每一天都在“祈祷”有个心仪的分析结果,终于在这天随着邮件提示音的响起,收到了分析结果......
分析前工作
爱基在进行数据分析之前,会有两次质控报告反馈给老师们。第一个,基因组DNA的提取质控报告(图1):保证DNA的完整性以及足够的量进行后续的富集亲和纯化;第二个,富集建库报告:构建DNA文库,利用磁珠富集与加完halo Tag标签表达的目的蛋白结合DNA片段,并纯化获得IP文库。这个过程中,为了检测蛋白表达的正常,爱基利用抗体对富集产物进行 WB 检测,同样对于文库也会进行质检(图2)。
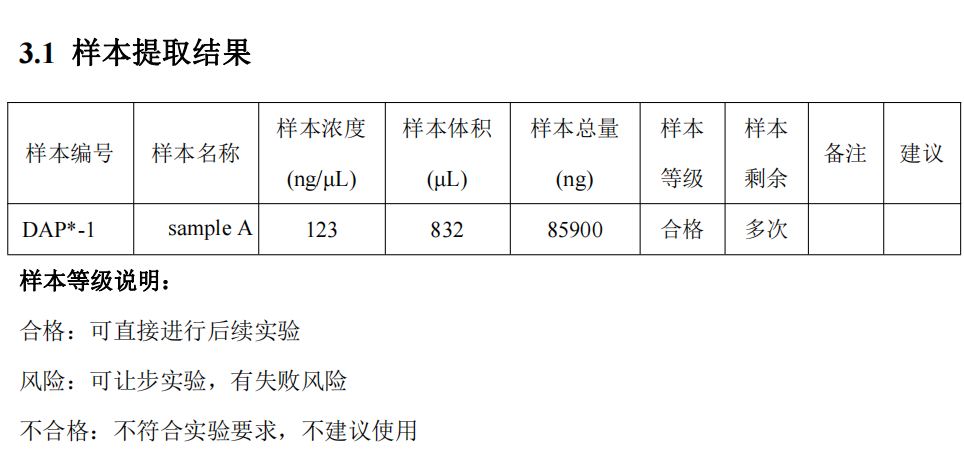

图1 DNA提取质控报告

图2 WB结果显示目的蛋白表达正常
 最低0.47元/天 解锁文章
最低0.47元/天 解锁文章


















 567
567

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









