











研究背景
碱性交换膜燃料电池受到阳极氢氧化反应(HOR)动力学缓慢的阻碍。近年来,异质结构催化剂在调整电子结构以促进催化性能方面显示出了巨大的潜力。考虑到Ni与TMNs的异质化具有很大的电催化潜力。
在此,中国石油大学赵联明和徐静等人通过密度泛函理论计算,系统地研究了Ni与过渡金属氮化物(Ni/TMNs、TMN=δ3−MoN、δ1−MoN和WN)异质结构的碱性HOR性能。计算方法在这项工作中,通过Materials studio量子计算软件包的Dmol3进行第一性原理理论计算,采用广义梯度近似法,以伯克-恩泽霍夫(PBE)泛函计算了交换相关能。
本文采用密度泛函半核伪势和双数值加极化基集分别来描述金属离子核和价电子函数,实空间全局轨道截止半径设置为5.1 Å,采用类导体屏蔽模型(COSMO)模拟溶剂环境。采用5×5×1Monkhorst-Pack网格进行布里渊区k点采样。此外,在计算态密度时,将k点的值增加到8×8×1,以保证精度。利用线性同步跃迁/二次同步跃迁法搜索了一个基本反应的过渡态,并通过振动频率分析进行了验证。同时能量和最大力移的收敛率分别设置为10-5 Ha和0.002Ha/Å,最大位移为5.0×10-3Å。
结果与讨论
为了探索TMN表面的稳定性,作者研究了N端和TM端TMN(0001)表面的表面能。如图1所示,所有TMNs的N端表面的表面能始终低于TM端表面,说明TMN(0001)表面更倾向于被N原子终止。这与Yuan的发现一致,其中δ1−MoN最稳定的表面是N端MoN(0001)。因此,通过将Ni纳米线加载到TMN(0001)衬底的N端表面,构建了Ni/TMN异质结构。
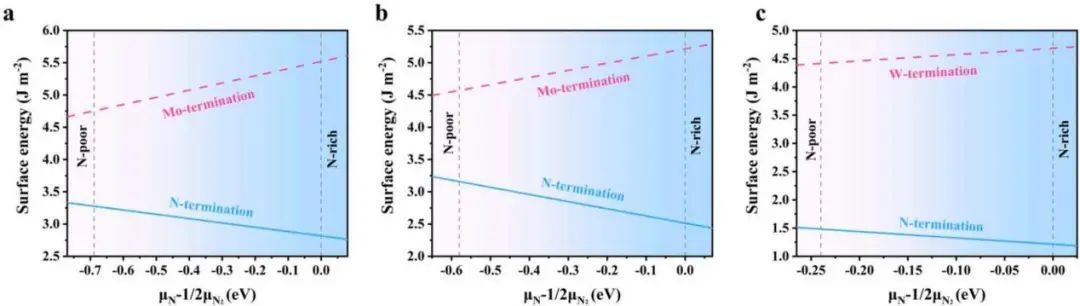
图1 TM端和N端材料的表面能与化学势的关系
Ni/δ3−MoN、Ni/δ1−MoN、Ni/WN的构型如图2所示。考虑到Ni和TMNs晶格参数不匹配导致的不可避免的应变,计算了Ni/TMNs和Ni(111)之间的平均Ni-Ni键长(d(Ni-Ni))的差异。如表S2所示,与纯Ni(111)(2.453Å)相比,Ni/δ3−MoN(2.408 Å)、Ni/δ1−MoN(2.438 Å)和Ni/WN(2.403Å)中的d(Ni-Ni)分别略微降低了1.8%、0.6%和2.1%。这表明Ni纳米线与TMN的匹配良好。虽然异质结构中支撑的镍纳米线略有变形,但整体结构保持稳定。
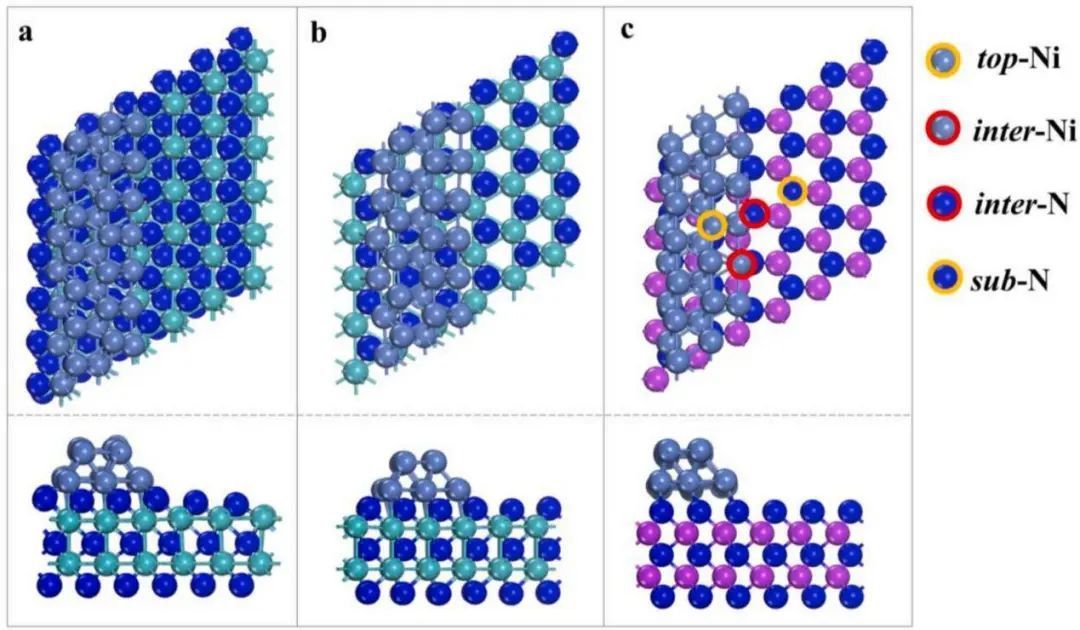
图2 催化剂结构图
为了进一步验证结构的稳定性,计算了TMN负载的Ni纳米线、孤立的Ni纳米线和Ni体的内聚能(见图3a)。如图3a所示,负载TMN的Ni纳米线的内聚能(5.00 ~ 5.21 eV)明显高于分离的Ni纳米线(3.80 eV)和Ni体(4.44 eV),表明Ni纳米线更倾向于锚定在TMN基底上,而不是扩散或聚集。
为了探索Ni/TMN异质结构的电子传递性质,计算了Ni和TMNs的功函数。如图3b所示,计算出Ni的功函数为5.09 eV,与之前报道的值很接近。而δ3−MoN、δ1−MoN和WN的功函数分别为7.24、7.43和7.54 eV。因此,Ni/TMNs中的电子倾向于从具有低功函数的Ni转移到具有高功函数的TMNs。通过Hirshfeld电荷和变形电子密度(DED)分析进一步证实了这一情况。
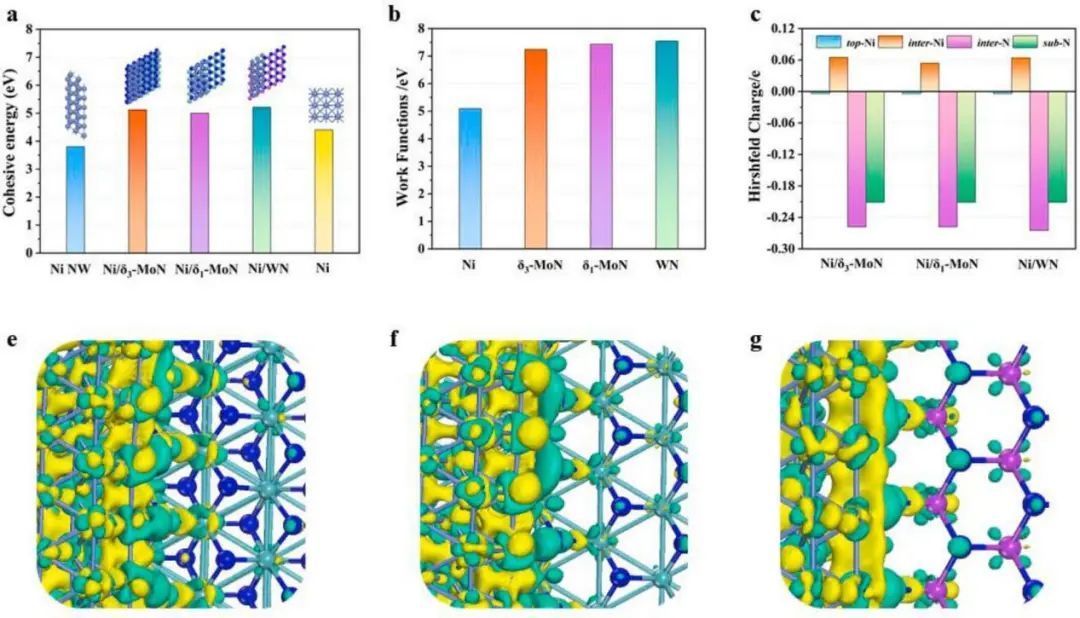
图3 异质结构内聚能,功函数及电荷变化
作者计算了投影态密度(PDOS),进一步探索了异质结界面间的电子相互作用。如图4a-c所示,与单相TMNs相比,Ni/δ3−MoN、Ni/δ1−MoN和Ni/ WN的异质结界面中n间的p带中心(εp)分别负移为−4.37、−3.96和−3.70 eV。相应地,Ni/TMNs中费米能级以上的未占据p态小于TMNs,这将导致吸附物的吸附较弱。另一方面,Ni/δ3−MoN的Nid带中心(εd)、Ni/δ1−MoN和Ni/WN之间分别为−1.70、−1.59和−1.61eV(见图4d)。Ni/TMNs中Ni d带中心的上移表明,有更多的未被占用的三维轨道来接受吸附物的电子,这将提高与吸附物的结合。
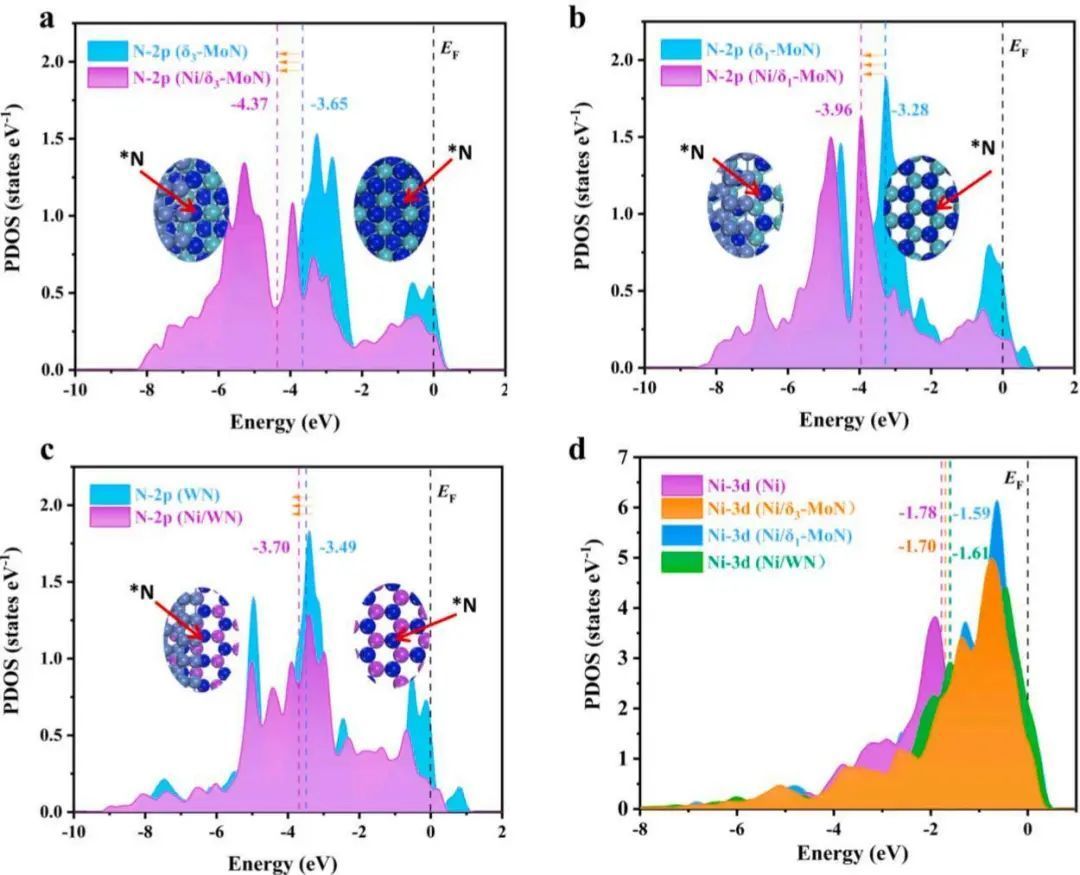
图4 催化剂态密度
为了探究异质结构上增强吸附的起源,绘制了Ni和Ni/TMNs上OH*吸附态的轨道耦合态图。如图5所示,OH*的O-2p态与Ni/TMNs的Ni-3d态的电子云符合度明显大于单相Ni,表明成键轨道填充增加,这是OH*吸附增强的原因。因此,OH*吸附来自于Ni/TMN界面上的相互调节的电子结构。
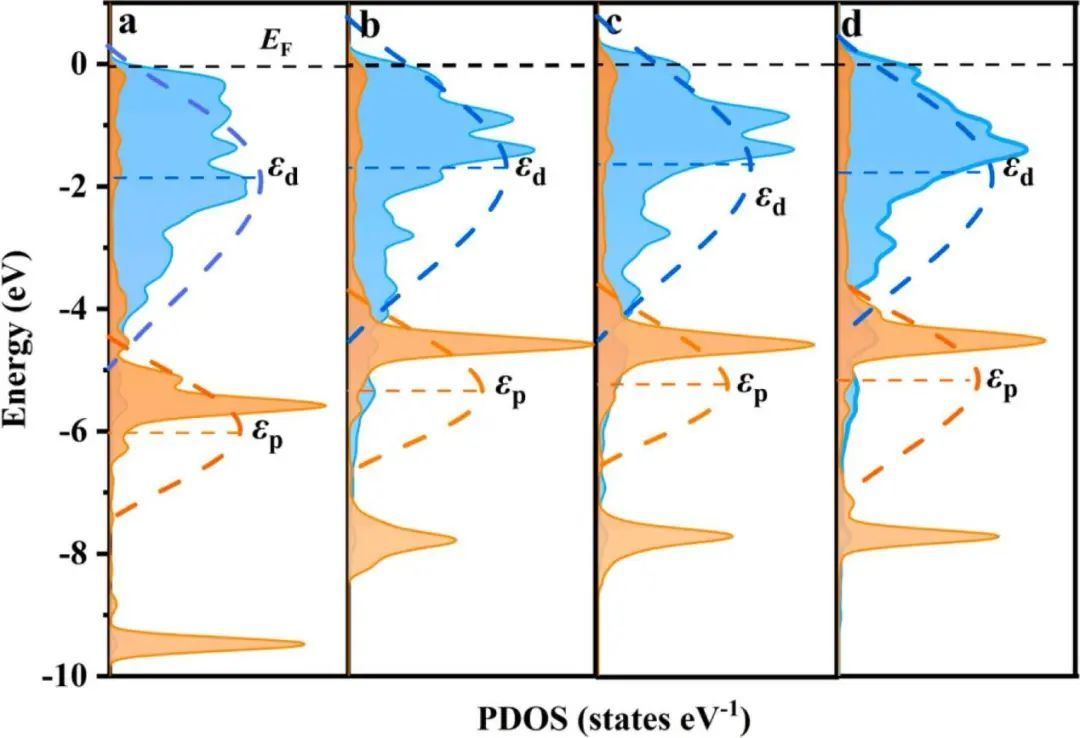
图5 吸附OH*时的催化剂态密度
对于碱性燃料电池,碱性介质中含有大量的氢氧化物物质,它们会在Ni(111)和TMN(0001)表面以及Ni/TMN界面上形成吸附的OH*态。图6a显示了氢氧化物吸附的自由能图。氢氧化物→OH*+e−)。在TMN催化剂上,氢氧化物物种经0.15 ~0.23 eV的吸热形成OH*,但在Ni上成为放热过程(ΔG =−0.14 eV)。在Ni/TMNs上,氢氧化物在Ni间位置吸附释放的热量进一步增加到−0.38 ~−0.46 eV,表明吸附的OH*很容易在异质结界面上形成。
因此,本文讨论了羟基作为吸附OH*态的HOR机理。H2的直接解离可以通过Tafel机制产生两个吸附的H*。在这个过程中,分子H2在Ni/TMNs上Ni间位点的拉伸振动使两个H原子相互分离,并分别转移到两个相邻的N间位点.如图6b所示,Ni/TMNs的自由能垒仅为0.12 ~ 0.33 eV,略高于Ni(0.06 eV),但明显低于TMNs的自由能垒(0.43 ~ 0.78 eV)。H2也可以与吸附的OH*反应形成吸附的H*并释放水分子。
最初,H2*和OH*位于Ni/TMNs上两个相邻的Ni间位点。然后,随着H−H键的延伸,H2*的一个H原子与相邻的OH*结合形成水分子,另一个H逐渐转向N间位。与Tafel机理相比,该过程需要克服Ni/ TMNs的0.34~0.64eV,Ni的0.44eV,TMNs的1.01 ~ 1.39 eV。
水分子可以通过H*和OH*的重组而形成。在Ni/TMNs上,Ni间位点的H*物种逐渐向邻近的N间位点的OH*物种移动,形成一个水分子。如图6d所示,由于H*在异质结和OH*在异质结界面上的吸附能优化,Ni/TMNs(0.49 ~ 0.72 eV)远低于Ni(1.15 eV)和TMNs(1.55 ~ 1.83 eV)。
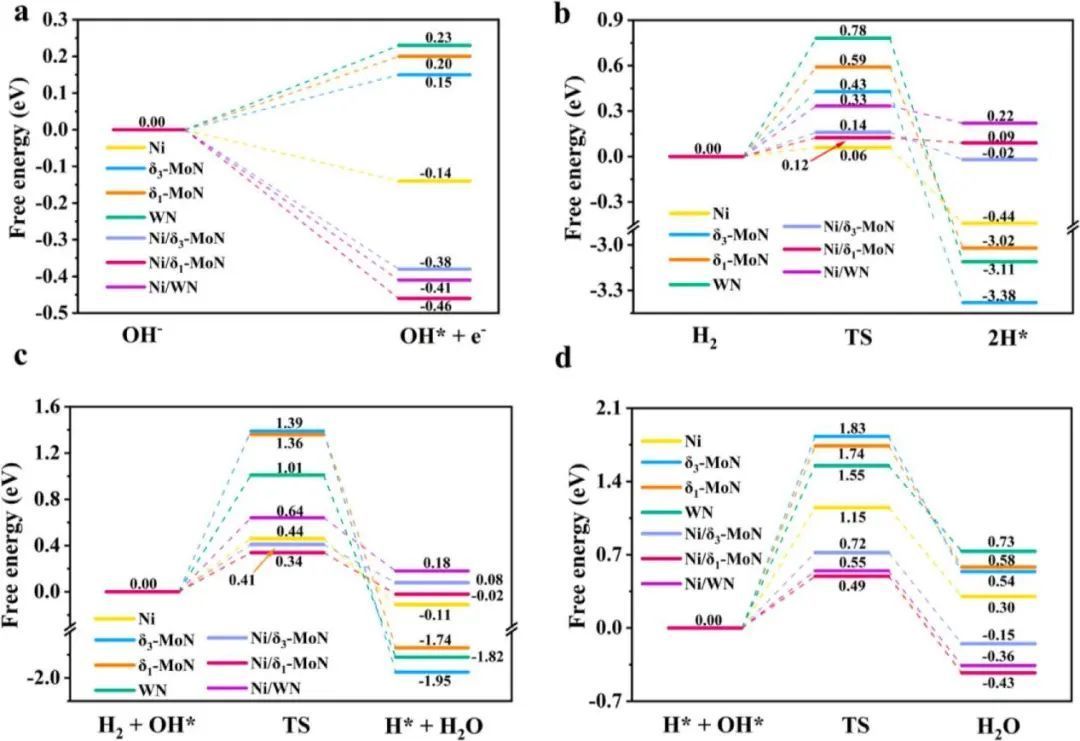
图6 催化剂的HOR反应自由能
为了了解H*和OH*吸附与HOR活性之间的关系,作者绘制了RDS(即H*+OH*反应生成H2O)的自由能垒(Ea)分别作为H*吸附自由能(ΔGH*)和OH*吸附自由能(ΔGOH*)的函数。如图8a所示,Ea值随着|ΔGH*|的增加而线性增加(Ea = 0.71|ΔGH*| + 0.57, R2 = 0.92)。
因此,|ΔGH*|的值越接近于零,自由能势垒越低。这与先前的研究一致,其中H原子与催化剂的相互作用强度具有最大化HOR活性的最佳值。同时,Ea与ΔGOH*呈线性正相关(Ea = 1.84ΔGOH* + 1.37, R2 = 0.94),说明自由能垒随着OH*吸附的增强而降低。与TMN和Ni相比,Ni和TMN的异质化可以有效地增强OH*的吸附,从而促进HOR。
在这项工作中,可以推断出所有TMNs, Ni和Ni/TMNs的ΔGOH*都位于最优值的弱吸附侧,从而导致HOR活性随着ΔGOH*的增强而增加。
此外,还发现HOR活性最高的Ni/δ1−MoN的H2O*吸附自由能值适中(与催化剂结合不太强也不太弱)。对于TMNs而言,H*结合强度(−1.57 ~−1.78 eV)太强,OH*吸附强度(0.15 ~ 0.23 eV)较差,导致H*和OH*复合的能垒过高,导致HOR的无催化活性。
与TMNs相比,Ni的H* + OH*反应的能垒明显降低,因为H*结合减弱(- 0.37 eV), OH*吸附增强(- 0.14 eV)。Ni与TMNs异质化后,界面处更加热中性的ΔGH*(−0.19 ~ 0.14 eV)和进一步增强的ΔGOH*(−0.38 ~−0.46 eV)非常有利于H*和OH*的复合,从而表现出较高的HOR活性。
特别是Ni/δ1-MoN具有几乎热中性的ΔGH* (-0.09 eV)和最强的ΔGOH* (-0.46 eV),解释了最低的HOR能垒。H*和OH*在Ni/TMNs上的最佳吸附源于界面上的双活性位点。在异质结界面处,Ni和TMNs之间的协同电子效应有效调节了H*和OH*在N间和Ni间的结合强度(见图3),通过双功能机制加速了HOR过程中H*和OH*的重组。
此外,如图8c所示,构建了2.5ΔGOH* + |ΔGH*|与HOR活性的线性关系图(Ea = 0.37 × (2.5ΔGOH* + |ΔGH*|) + 0.96, R2 = 0.95),表明HOR活性对OH*吸附强度更为敏感。因此,与H*吸附相比,改变OH*吸附能力可能更有效地提高Ni/TMN催化剂的HOR性能。

图7 决速步骤标度关系
结论与展望
综上所述,通过DFT计算研究了Ni/TMN异质结构催化剂的HOR性能。Ni/ TMNs上的HOR主要通过Tafel反应发生,然后是H*和OH*重组的速率决定步骤。本研究为合理设计针对AEMFCs的高效镍基HOR催化剂提供了广阔的应用前景。
文献信息
Zhao, L., Tong, Y., Ding, Y., Kong, W., Wang, J., Li, B., … & Xing, W. (2023). Designing interface structures of nickel with transition metal nitrides for enhanced hydrogen electro-oxidation. Surfaces and Interfaces, 102659.















 4333
4333











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









