







 文章讨论了小样本量实验设计的四种类型,包括发育周期、跨器官比较、野生型与突变体对比和处理时间/浓度梯度,以及大样本量下的农学和医学研究方法,如群体研究、队列研究和泛癌分析。通过案例研究展示了转录组和表观组学在不同领域的应用,强调了实验设计对实验成功的重要性。
文章讨论了小样本量实验设计的四种类型,包括发育周期、跨器官比较、野生型与突变体对比和处理时间/浓度梯度,以及大样本量下的农学和医学研究方法,如群体研究、队列研究和泛癌分析。通过案例研究展示了转录组和表观组学在不同领域的应用,强调了实验设计对实验成功的重要性。

在进行测序实验之前,精心设计实验分组是实验成功的关键一步。
根据样本量差异,有不同的实验设计思路。今天我们以转录组为例,讲讲常见测序前的实验分组思路,分别从小样本量和大样本量谈谈农学和医学上的分组思路。
No.1 小样本量
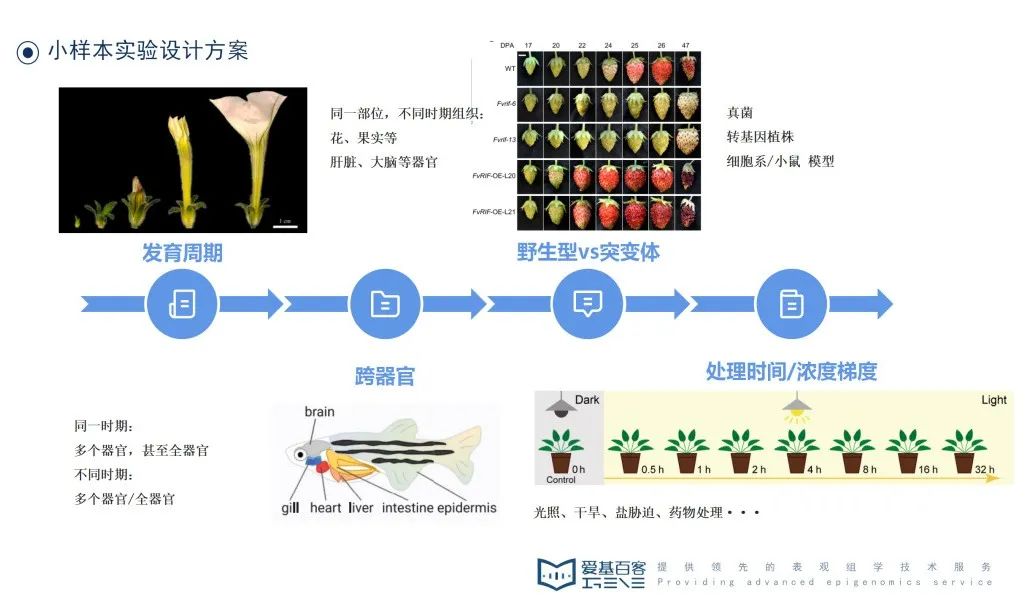
小样本量实验设计方案主要有4种类型:发育周期、跨器官、野生型vs 突变体、处理时间/浓度梯度。
-
发育周期:这一类研究通常适用于研究器官发育、果实成熟的研究,取样尽量遵循同一部位,剔除其他组织。如何确定时间节点?可以通过文献资料、植物学手册、数据库和实验观察等设计取样时间点。
-
跨器官:选取同一时期的多个器官甚至全器官,或选不同时期多个器官/全器官。这一类研究通常用于构建物种的器官图谱。
-
野生型vs突变体:当我们手上有一个待研究基因,为了确定其功能,通常会构建相关突变体。进行测序时,选取表型差异明显的样本,通常也会利用RT-PCR确定基因超表达/沉默的效果。
-
处理时间/浓度梯度:这一类研究要么有具体的研究条件(干旱、盐、光照等),适合逆境胁迫的研究,或者是药物一类的处理。关于处理时间和浓度梯度上,建议提前做好预实验,有较为明显差异的表型,再准备样本。
案例一:时间处理梯度 | 表观组+转录组解释番木瓜果实成熟机制
ATAC-seq and RNA-seq reveal the role of AGL18 in regulating fruit ripening via ethylene-auxin crosstalk in papaya. 2022
发表期刊:Postharvest Biology and Technology
研究单位:华南农业大学
研究技术:转录组+表观组
样本:9个
研究内容:
木瓜(Carica papaya)是第三大产热带水果。它是一种气味素果,因生理迅速分解而极易腐烂。化合物1-MCP是一种有效的乙烯受体抑制剂,可以与乙烯受体相互作用,避免其生理激活。
研究对1-MCP处理后果实2h和16h的样本进行ATAC-seq和RNA-seq测序,分析差异开放区域和差异表达基因。整合分析ATAC-seq和RNA-seq数据,建立了短期和长期1-MCP处理下与成熟相关基因的表达谱。研究发现多个转录因子,包括MADS(CpAGL18),以及八个与果实软化和成熟相关的差异表达基因(DEGs)。CpAGL18表达在控制条件下成熟期间显著增加,但在长期1-MCP处理下被强烈抑制。启动子分析预测了MADS在CpACS1和CpSAUR32中的顺式作用元件。此外,EMSA、Y1H和ChIP-qPCR实验证实了CpAGL18在体内和体外结合到CpACS1和CpSAUR32启动子上。最后,双荧光素酶报告基因实验证明CpAGL18激活了CpACS1和CpSAUR32的启动子。这些发现表明,CpAGL18通过整合乙烯和生长素信号,在木瓜的果实软化和成熟中发挥关键作用。
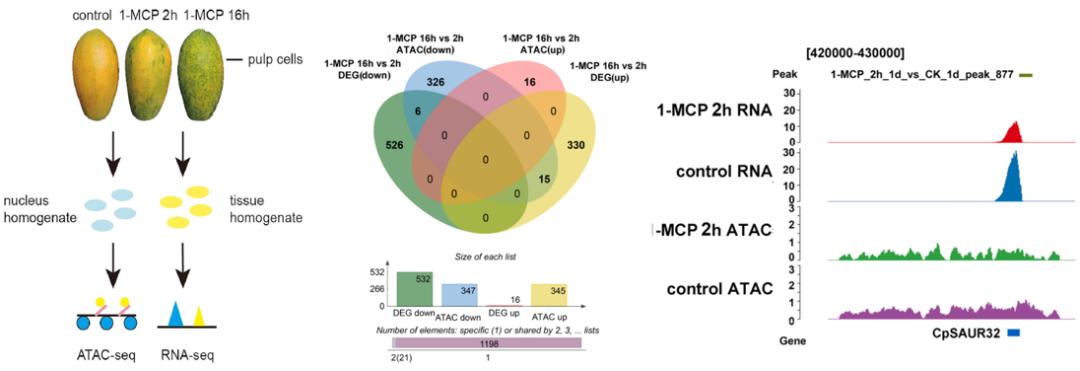
图:实验设计,ATAC-seq+RNA-seq联合分析。
案例二:发育周期 | 转录组+代谢组解析猕猴桃的主要品质调控机制
A comprehensive metabolic map reveals major quality regulations in red-flesh kiwifruit (Actinidia chinensis). 2023
发表单位:New Phytol
研究单位:四川大学
研究技术:转录组+代谢组
样本:33
研究内容:
猕猴桃是全球受欢迎的水果之一,其品质主要由关键代谢物(糖类、类黄酮和维生素)决定。以往对猕猴桃的研究大多采用单一组学方法,或仅涉及有限的代谢物。因此,猕猴桃在发育和成熟过程中的动态代谢组和潜在调控机制尚不明确。在这项研究中,通过代谢组学和转录组学分析(猕猴桃11个不同发育和成熟阶段),将515种代谢物及其共表达基因平行分类为10个不同的代谢vs基因模块。通过整合生物信息学和功能基因组学实验,研究构建了全局图谱,并揭示了贯穿猕猴桃生长周期的所有主要代谢变化的基因组和转录调控网络。除了已知的代谢vs基因模块如可溶性糖类外,研究还发现了调控原花青素、维生素C和其他重要代谢物积累的新型转录因子。总之,研究发现揭示了猕猴桃代谢调控网络,并为改善猕猴桃品质提供了宝贵资源。
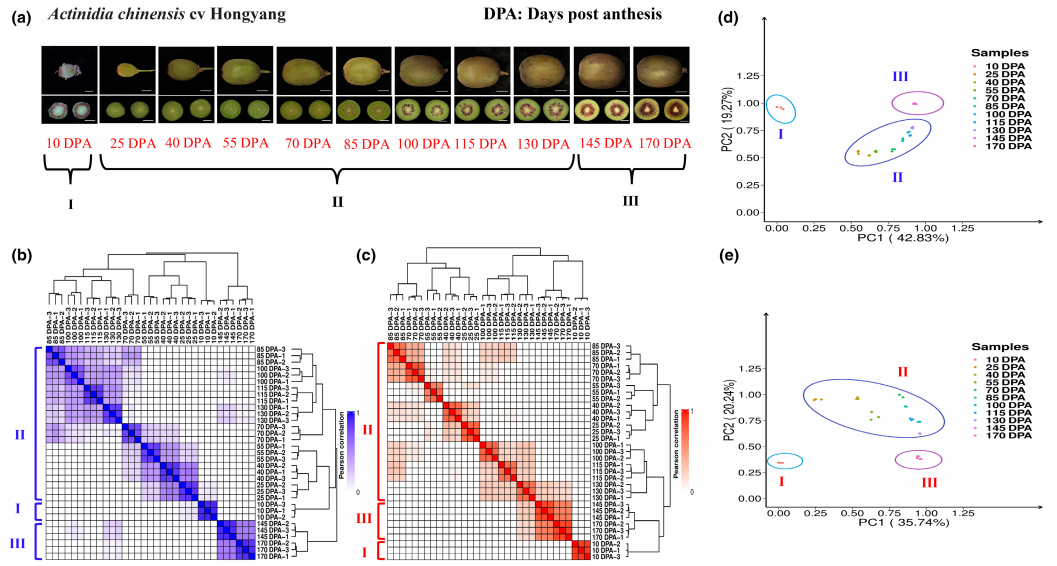
图:代谢组+转录组数据集。
案例三:发育周期 | 鸡卵泡形成过程中颗粒细胞的动态转录组和染色质结构
Dynamic transcriptome and chromatin architecture in granulosa cells during chicken folliculogenesis. 2022
发表期刊:Nature Communications
研究单位:四川农业大学
研究技术:转录组+表观组(ChIP-seq、Hi-C、ATAC-seq)
样本:60
研究内容:
研究调查了鸡卵泡发生过程中颗粒细胞的转录组动态(10个时期,每个时期6个生物学重复),并评估了染色质结构动态及其在三个关键阶段(小白卵泡前阶段、第一个最大的排卵前卵泡和排卵后卵泡)对颗粒细胞基因表达的影响。结果显示染色质结构全局重编程与转录组差异之间的一致性,提供了区段重排、拓扑结合域的可变组织和启动子与增强子之间长程相互作用的重组证据。这些结果为鸟类生殖生物学提供了重要见解,并为未来对颗粒细胞进行深入功能表征提供了基础数据集。
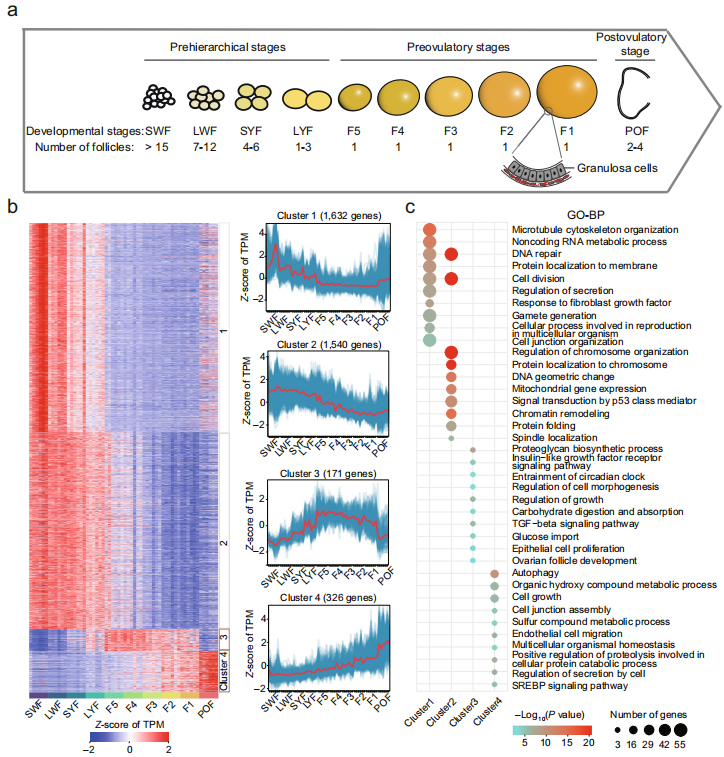
图:卵泡发生过程中颗粒细胞(GCs)的转录组谱。
案例四:对照vs突变体 | 转录因子BATF的耗竭增强了CAR-T细胞对实体肿瘤的抗肿瘤活性
Depletion of BATF in CAR-T cells enhances antitumor activity by inducing resistance against exhaustion and formation of central memory cells.2022
发表期刊:Cancer Cell
研究单位:中国科学院动物学研究所
研究技术:转录组+表观组
研究内容:
嵌合抗原受体(CAR)T细胞疗法对实体瘤的效果有限,其中一个主要挑战是T细胞耗竭。为了应对这一挑战,研究使用一个低功能CAR-T细胞模型进行了候选基因筛选,并发现基本亮氨酸拉链ATF样转录因子(BATF)的耗竭改善了CAR-T细胞的抗肿瘤性能。在不同类型的CAR-T细胞和小鼠OT-1细胞中,失去BATF赋予T细胞更好的耐耗竭性和更高的肿瘤根除效果。从机制上,研究发现BATF与人类CAR-T细胞中一组与耗竭相关的基因结合并上调其表达。BATF调控参与效应和记忆T细胞发展的基因表达,敲除BATF使细胞群体向更中央记忆亚群转变。研究证明了BATF是限制CAR-T细胞功能的关键因素,其耗竭增强了CAR-T细胞对实体瘤的抗肿瘤活性。
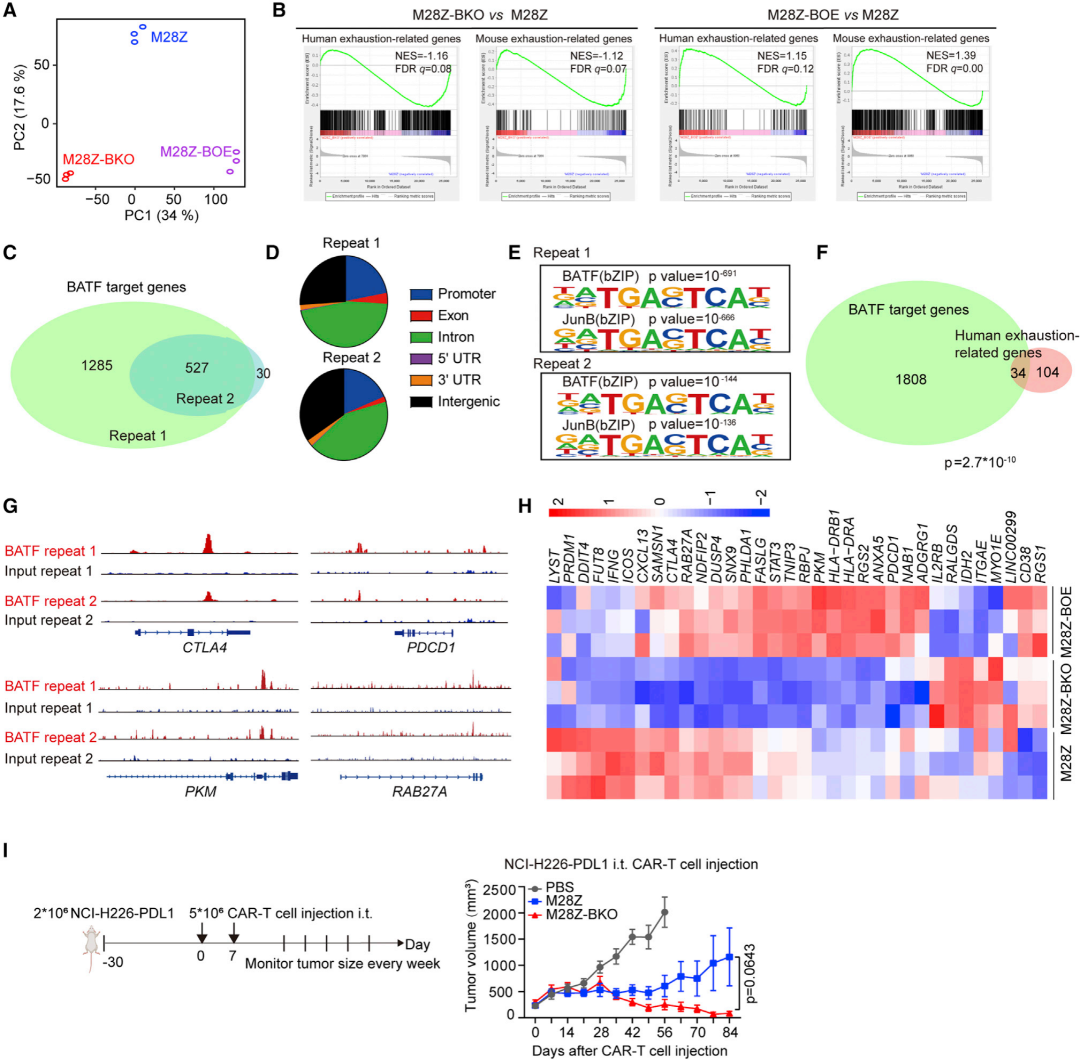
图:RNA-seq和ChIP-seq数据集:BATF导致CAR-T细胞衰竭。
No.2 大样本量
针对大样本量组学实验设计上农学和医学上有差异,农学上主要有群体研究、跨/全器官图谱、泛物种+多器官等,医学上大样本量研究主要有队列研究和泛癌分析。

-
群体研究
在农学上,群体遗传研究主要聚焦于作物、畜牧和水产等农业生物群体的遗传多样性、遗传结构以及遗传变异对于农业性状的影响。这类研究对于作物改良、种质资源保护、疾病防控和可持续农业发展具有重要意义。
案例五:eQTL在调控水稻基因表达和鉴定关键调控因子中发挥着关键作用
eQTLs play critical roles in regulating gene expression and identifying key regulators in rice. 2022
发表单位:Plant Biotechnology Journal
研究单位:华中农业大学
研究技术:转录组
样本:287个
研究内容:
在这项研究中,对来自世界各地的287个稻米品种的叶片转录组进行了测序,并获得了总共177,853个高质量的单核苷酸多态性(snp)。基因组范围关联研究确定了44,354个表达数量性状位点(eQTLs),调控着13,201个基因的表达,以及17个局部eQTL热点和96个远端eQTL热点。此外,转录组范围关联研究筛选出了在拔节期叶片淀粉含量中的21个候选基因。HS002被确定为一个重要的远端eQTL热点,下游有五个富集于二萜抗毒素合成的基因。共表达分析、eQTL分析和连锁图谱分析共同证明了bHLH026作为一个关键调节因子,激活了下游基因的表达。转基因实验揭示了bHLH026是二萜抗毒素合成的重要调节因子,并增强了稻米的抗病能力。
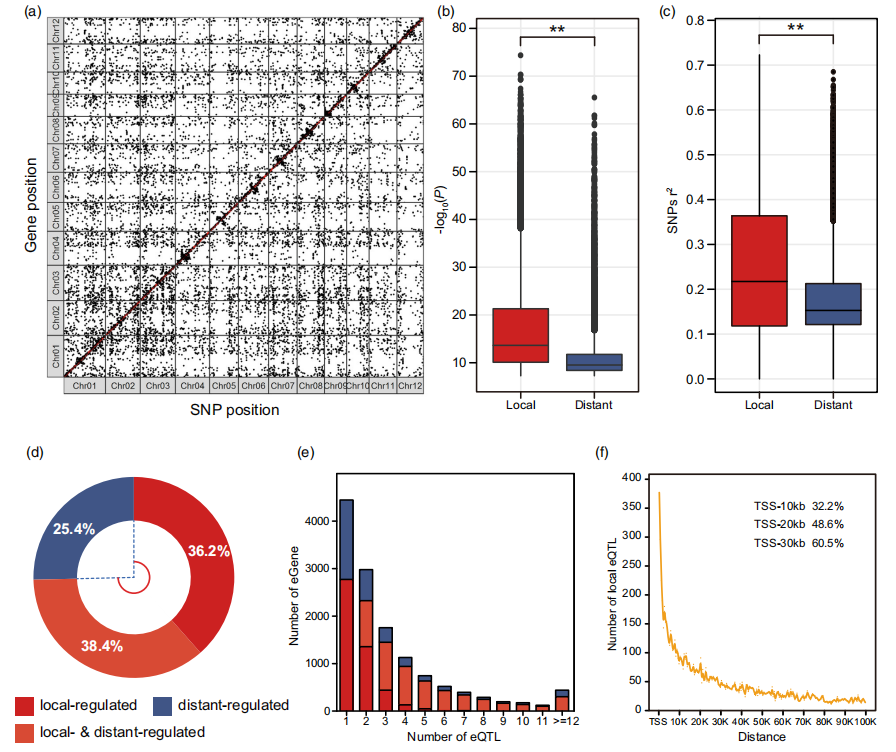
图:利用抽穗期水稻旗叶的RNA-Seq数据鉴定eQTL。
-
多器官
对多器官的大规模组学分析,通过对多个器官的组学数据(比如基因组、转录组)进行比较和分析,可以揭示不同器官之间的遗传差异、基因表达模式的变化以及器官间相互作用的复杂性。这种综合性的研究方法有助于揭示生物体内复杂的生物学过程,为理解生物体的整体功能和适应性提供重要见解。
案例六:大规模的基因组学和转录组学分析阐明了鸡的高肉产量的遗传基础
Large-scale genomic and transcriptomic analyses elucidate the genetic basis of high meat yield in chickens. 2023
发表期刊:Journal of Advanced Research
发表单位:中国农业科学院动物科学研究所
研究技术:基因组+转录组
样本:129(转录组)
研究内容:
对3只纯种肉鸡(n = 748)和6个地方品种/系(n = 114)进行了全基因组重测序,并从NCBI数据库中获得了12个鸡品种(n = 199)的测序数据。此外,对两个鸡品种(n = 129)在两个发育阶段的6个组织进行了转录组测序。采用顺式eQTL定位和孟德尔随机化相结合的全基因组关联研究。研究构建了一个包含与肌肉发育相关的多个组织和阶段的基因组变异和转录组变异的大规模图谱,为探索肉鸡生产性状的遗传基础提供了充分和有价值的信息。根据基因组和转录组学证据,胸肌是纯种肉鸡与当地鸡或野生祖先鸡GGS的主要差异。
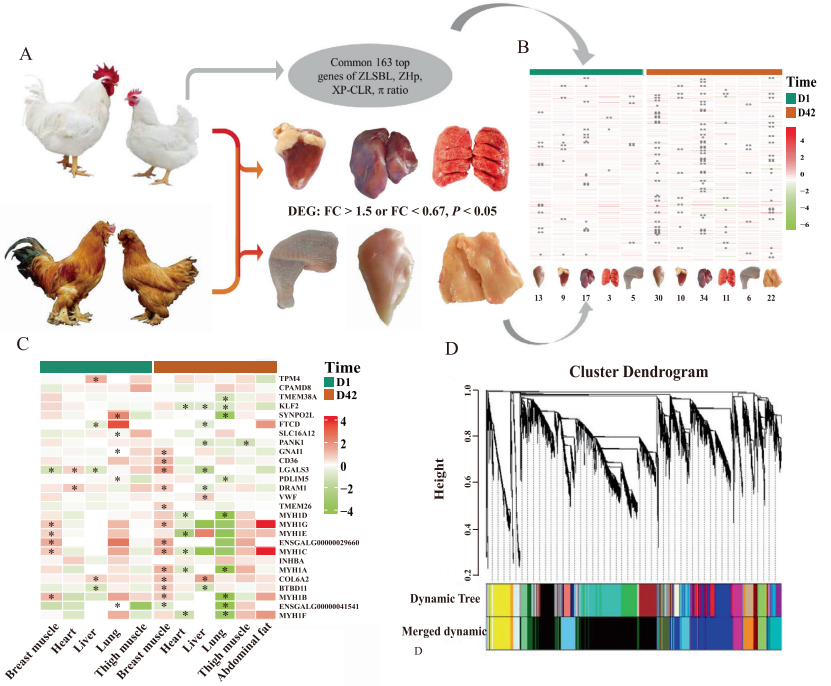
图:纯种肉鸡B系和本地品种BJY鸡的推测选择基因(PSGs)的转录组学图谱。
-
泛物种+多器官
案例七:比较转录组分析揭示了支撑陆地植物器官发生和繁殖的保守程序。
Comparative transcriptomic analysis reveals conserved programmes underpinning organogenesis and reproduction in land plants. 2021
发表单位:Nature Plants
研究单位:南洋理工大学
研究技术:转录组(比较转录组)
样本:4806个(从SRA数据库中下载了4672个RNA-seq数据,自测134个RNA-seq样本)
研究内容:
植物器官的出现引发了陆地植物的爆发性辐射,塑造了生物圈并促成了陆地动物生命的建立。器官和固定配子的进化需要协调获得新基因功能、利用现有基因以及发展新的调控程序。然而,对于陆地植物尚未进行大规模的基因组和转录组数据分析。为了弥补这一缺陷,研究为包括苔藓植物、维管植物、裸子植物和被子植物在内的十种植物物种生成了各种器官和配子的基因表达图谱。对这些图谱的比较分析确定了数百个器官和配子特异性正交组,并揭示了大部分特异转录组的显著保守性。研究结果表明,利用现有基因是进化新器官的主要机制。与雌配子相比,雄配子显示出大量特定基因的高度保守性,这表明雄性生殖高度专门化。捕捉花粉发育的表达图谱揭示了许多对花粉生物发生和功能至关重要的转录因子和激酶。
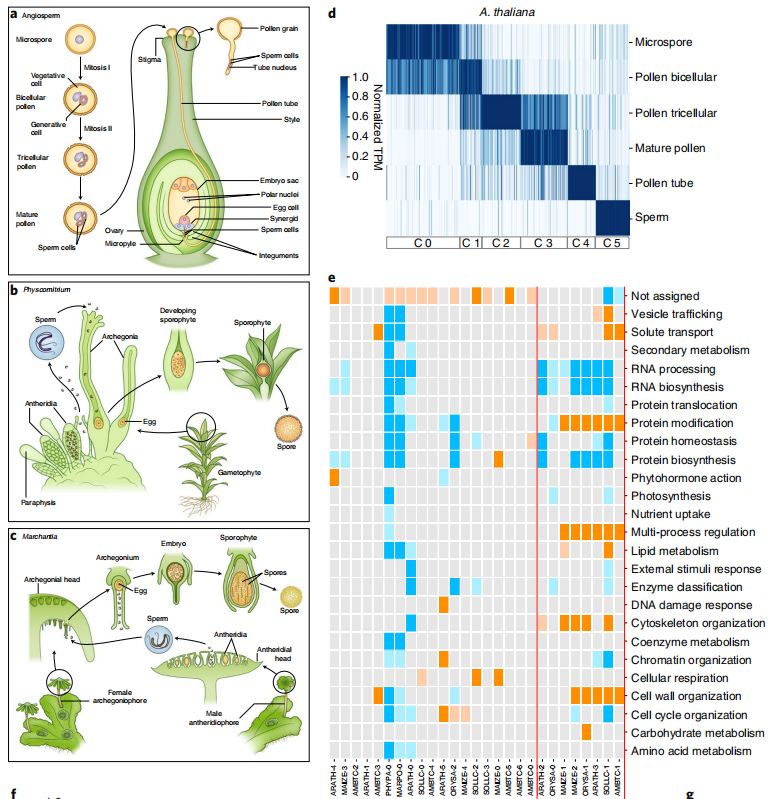
图:7种陆地植物的表达图谱。
-
队列研究
队列研究是一种观察性研究设计,广泛应用于医学和流行病学领域。它旨在评估暴露因素与健康结果之间的关系。队列研究是探讨疾病病因的常用方法之一,其论证强度较高,能较好地揭示两事件间客观存在着的因果关系。常常用在疾病预后、临床试验等研究中。
较为常见大队列组学研究有:肿瘤队列、出生队列、流行病疾病队列。
队列研究常用组学技术:重测序、外显子测序、转录组、表观组、代谢组和蛋白组。
为什么做队列研究?我们在临床研究中发现,相同的治疗方案经常会对个体产生不同的疗效。举个例子:相同的治疗药物,对部分患者可能会产生比较好的疗效,但是对其他患者可能无效甚至会加重疾病进展。这些现象涉及到疾病发病机制、诊断评估、药物疗效、生存预后等个体差异。队列研究正适合解决这些问题。
案例八:哮喘气道转录组全关联研究揭示了遗传驱动的粘液病理生物学
Nasal airway transcriptome-wide association study of asthma reveals genetically driven mucus pathobiology.2022
发表期刊:Nature Communications
研究单位:National Jewish Health
研究技术:全基因组+转录组
样本:695
研究内容:
为了确定气道功能障碍的遗传因素,研究通过将681名儿童鼻气道上皮的RNA-seq数据与英国生物库的遗传关联数据相结合,进行了哮喘的转录组范围关联研究。气道分析确定了102个哮喘基因,其中58个在使用其他哮喘相关组织进行的转录组范围关联分析中未被发现。这些基因包括MUC5AC(气道粘蛋白)和FOXA3(黏液转化的转录驱动因子)。基因分型供体的粘膜纤毛上皮培养显示,MUC5AC风险变异体增加了MUC5AC蛋白分泌和黏液分泌细胞频率。针对黏液产生和慢性咳嗽的气道转录组范围关联分析也确定了MUC5AC。这些顺式表达变异体与转录效应相关联;MUC5AC变异体与非炎症性黏液分泌网络基因的上调相关,而FOXA3变异体与上调第二型炎症诱导的黏液转化途径基因相关。总的来说,该研究结果揭示了气道黏液病理生物学的遗传机制。
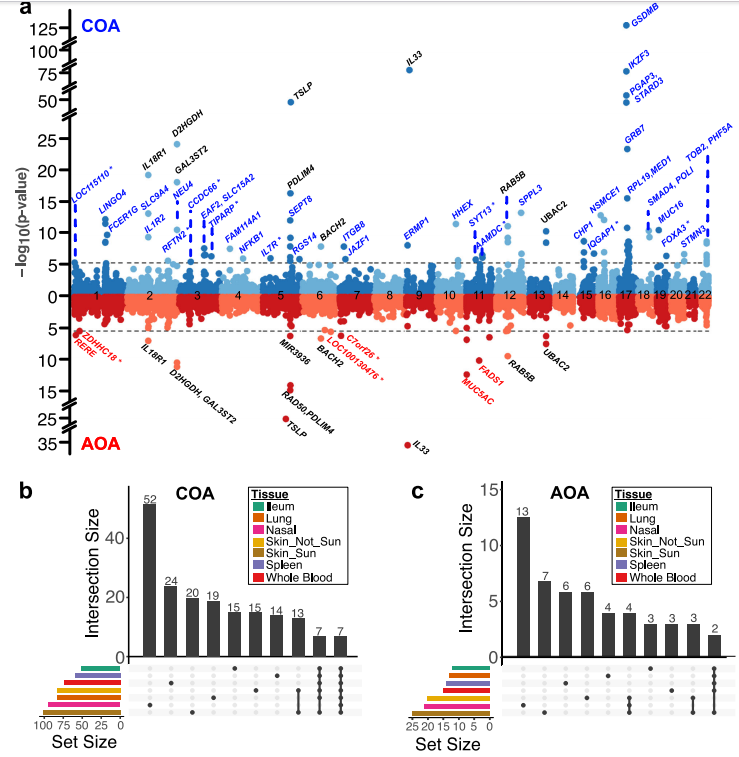
图:鼻气道上皮TWAS(transcriptome-wide association study)识别组织特异性COA/AOA基因。
-
泛癌分析
Pan-cancer分析是一种全面的研究方法,旨在跨越多种癌症类型,通过比较和对比不同癌症之间的共性和差异性,来发现癌症的普遍特征和潜在治疗靶点。这种研究方法通过整合和分析来自多种癌症的大量生物信息学数据,尝试揭示癌症发生和发展的共同机制,以及寻找可能对多种癌症有效的治疗方法。
Pan-cancer研究通常多种组学都有涉及:基因组学、转录组学、蛋白质组学、代谢组学、表观组学、单细胞测序。泛癌分析通常会与队列研究结合。
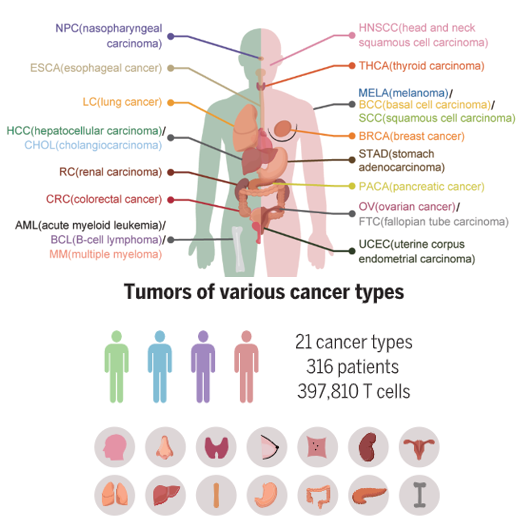
Zheng et al.,Science374, 1462 (2021)
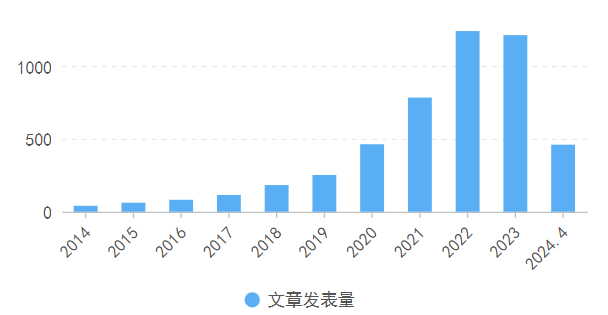
利用pubmed检索pan-cancer,可以看到近三年相关研究发表量逐年走高。
案例九:难治性转移性癌症的整合泛癌基因组学和转录组学分析
Integrative Pan-Cancer Genomic and Transcriptomic Analyses of Refractory Metastatic Cancer. 2023
发表单位:Cancer Discovery
研究单位:巴黎-萨克雷大学
研究技术:外显子测序+转录组
样本:947(转录组)+569(WES)
研究内容:
治疗后的转移性复发是导致癌症死亡的主要原因,对大多数患者接受的治疗缺乏已知的耐药机制。为弥补这一差距,研究分析了一个全癌症队列(META-PRISM),包括1,031个难治性转移性肿瘤的全外显子组和转录组测序数据。META-PRISM肿瘤,特别是前列腺、膀胱和胰腺类型,与未经治疗的原发性肿瘤相比,显示出基因组的最大转变。仅在肺癌和结肠癌中发现了标准治疗耐药生物标志物——META-PRISM肿瘤中的9.6%,表明太少的耐药机制已经得到临床验证。相反,研究验证了与未接受治疗的患者相比,接受治疗的患者中多种调查和假设的耐药机制的丰富化,从而确认了它们在治疗耐药性中的潜在作用。此外,研究证明了分子标志物可以改善6个月生存预测,特别是对于晚期乳腺癌患者。研究的分析确立了META-PRISM队列在研究耐药机制和进行癌症预测分析中的实用性。
图:META-PRISM 队列的临床特征、测序数据、治疗史和结果
综上所述,小样本量实验设计方案涵盖了发育周期、跨器官、野生型与突变体、以及处理时间/浓度梯度等不同类型,我们在设计分组时根据自己的研究目的选择合适的分组。在大样本量组学实验设计中,农学领域注重群体研究、跨/全器官图谱、以及泛物种+多器官等研究方式,而医学领域则更加关注队列研究和泛癌分析等方法。
武汉爱基百客生物科技有限公司配备高通量测序仪DNBSEQ-T7、MGI2000等,可以为客户提供全基因组测序、全外显子测序、转录组测序、微生物组测序等测序服务(日产数据高达14T,满足大型基因组和人群队列项目),另外作为表观组学研究的先行者,我们也提供表观组(甲基化、ChIP-seq、CUT&Tag、ATAC-seq等)服务,如果您有任何产品需求,欢迎联系我们。

















 1117
1117

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









