










 成果简介
成果简介
单原子催化剂由于具有良好定义的金属单原子位(M SASs)所带来的优异催化性能而成为材料科学领域的前沿热点。
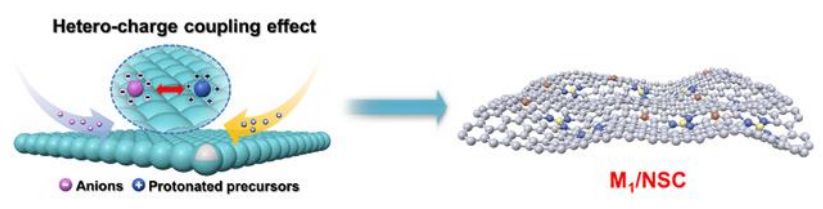
安徽工业大学吴孔林副教授、张奎教授、安徽工业大学/中科大曾杰教授等人报道了一种基于异电荷耦合效应(HCCE)的新型合成策略,以制备负载在N和S共掺杂多孔碳(M1/NSC)上的M SASs。该策略被广泛应用于制备17种单金属或多金属组成的配位环境和电子结构综合调控的M1/NSC,具有良好的通用性和灵活的可调节性。此外,该策略提供了一种低成本、高产能高效合成M1/NSC的方法,一次可生产50 g以上的催化剂,这是大规模生产的关键。
在所制备的多种一元M1/NSC催化剂(M可以是Fe、Co、Ni、V、Cr、Mn、Mo、Pd、W、Re、Ir、Pt或Bi)中,Fe1/NSC电催化硝酸盐还原为NH3的性能优异,NH3法拉第效率高达86.6%,NH3产率高达1.50 mg h-1 mgcat.-1(在-0.6 V下)。Fe1/NSC作为Zn-硝酸盐电池的正极材料,其开路电压高达1.756 V,能量密度高达4.42 mW cm-2,具有良好的循环稳定性。
相关工作以《A General Strategy Based on Hetero-Charge Coupling Effect for Constructing Single-Atom Sites》为题在《Angewandte Chemie International Edition》上发表论文。
值得注意的是,6月15日,中国化学会第34届学术年会开幕式在广州市白云国际会议中心举行,颁发中国化学会系列学术奖励。曾杰教授因“聚焦二氧化碳催化转化,提出催化剂设计新理念,开辟催化转化新路径”荣获“第十二届中国化学会-巴斯夫公司青年知识创新奖”,全国仅4人获此殊荣。

图文导读
 图1 Fe1/NSC的制备与表征
图1 Fe1/NSC的制备与表征
为了更好地了解HCCE的有效性和适用性,以Fe1/NSC为典型案例,阐述了HCCE的合成原理和过程。如图1a所示,分别以质子化苯胺、SiO2、K4[Fe(CN)6]和(NH4)2S2O8作为碳骨架和氮掺杂剂、成孔剂、金属阴离子源和引发剂。首先,用S2O82-对质子化苯胺进行聚合,将带正电的质子化苯胺结构单元和带负电的离子通过HCCE包封在聚苯胺(PANI)框架中。在氧化聚合过程中,部分S2O82-被还原成SO42-,形成S掺杂剂。搅拌24 h,室温下反应,离心得到SiO2@PANI-[Fe(CN)6]4-S2O82-/SO42-前驱体。最后,对冻干前驱体进行热解、蚀刻、氢活化,得到Fe1/NSC。值得注意的是,可以很容易地实现Fe1/NSC催化剂的规模化合成,一次产量可达53.33 g。
AC-HAADF-STEM图像显示Fe1/NSC的亮点对应于金属原子(图1b)。元素映射图显示Fe、N、S和C在Fe1/NSC上均匀分布,没有明显的纳米颗粒或团簇(图1c)。ICP-OES结果表明,Fe1/NSC中Fe的负载量为1.02 wt%。
此外,本文利用XAS进一步表征了原子分散的Fe位点的几何结构和化学环境。Fe1/NSC的Fe的K边缘XANES谱介于Fe箔和Fe3O4之间,表明Fe1/NSC中的Fe具有部分正电荷(图1d)。FT-EXAFS结果表明,Fe1/NSC主峰在1.45 Å处,没有Fe-Fe峰,证明Fe以原子分散状态存在(图1e)。随后,利用Fe1/NSC中Fe的K边缘FT-EXAFS谱来确定Fe的结构特征(图1f)。拟合结果表明,每个Fe原子与3个N原子配位形成Fe-N3结构单元。
 图2 各种M1/NSC的AC-HAADF-STEM图像
图2 各种M1/NSC的AC-HAADF-STEM图像
得到的Co1/NSC和Ni1/NSC进行了系统表征,以证明HCCE的可行性(图2a-2b)。通过AC-HAADF-STEM表征,在其他制备的M1/NSC中也观察到原子分散的M物种(图2c-2p)。进一步利用XRD、SEM、TEM、EDS元素图谱和XPS对制备的具有一元至五元金属位点的M1/NSC进行表征,确定M物种的存在状态和电子结构特征。这些结果表明,HCCE适用于可控和通用的M SASs合成,为未来扩大其应用领域提供了可能。
 图3 电催化NO3RR性能
图3 电催化NO3RR性能
为了评价合成的催化剂的性能,以电催化NO3RR为模型反应。图3a显示了使用M1/NSC催化剂将NO3-电催化转化为NH3的示意图。LSV曲线表明,与空白NSC相比,在NO3-存在下,Fe、Co、Ni、Cr和Mn1/NSC催化剂的电流密度显著增加(图3b)。如图3c所示,在不添加NO3-的情况下,NH3的核磁信号检测不到,只有添加Na14NO3或Na15NO3时,才会出现14NH3和15NH3的核磁峰,证实了NH3是通过电催化NO3RR由NO3-生成的。
相应的NH3法拉第效率和NH3产率如图3d所示。在-0.6 V的低电位下,Fe(86.6%)、Co(79.9%)、Ni(79.4%)、Cr(63.2%)、Mn(49.6%)和Bi1/NSC(28.2%)催化剂的NH3法拉第效率显著高于NSC(20.2%)催化剂。只有Fe1/NSC催化剂在更多电位下表现出稳定且较高的NH3法拉第效率(高于86.6%)。
采用原位ATR-FTIR技术捕获了NO3RR电催化过程中Fe1/NSC催化剂的反应中间体信息。如图3e所示,在-0.5到-1.1 V的电位范围内出现了几个反应中间体对应的峰。对于Fe1/NSC,观察到*NH2(1169 cm-1),*NH4+(1530 cm-1)和N-H弯曲(1666 cm-1)的特征峰,表明电催化NO3RR过程中产生NH3。
此外,本研究利用NSC确定NO3RR的电位决定步骤(PDS)为HNO*加氢生成HNOH*,自由能极高,达到1.94 eV(图3f)。高吸能的PDS伴随着HNO*与吡啶-N位点之间形成的环状结构的破坏。在NO3RR的早期阶段,HNO*与HNO2*和NO*在NSC上与吡啶-N和相邻的两个C原子形成五元环结构。这就是为什么吡啶-N比吡咯-N与这些中间体结合更强的原因。然而,当HNO*在O端进一步加氢时,没有形成环结构,反应能明显提高(图3f)。与NSC相比,利用Fe1/NSC的NO3RR的PDS为NO*加氢生成HNO*,反应自由能为0.83 eV,而其他基本反应均为放热的(图3g)。在U=-0.83 V时,Fe1/NSC的反应均能进行,这与实验结果一致,表明Fe1/NSC对NO3RR的催化性能优于NSC。
 图4 Zn-NO3-电池的性能
图4 Zn-NO3-电池的性能
为了探索该催化剂在实际应用中的可能性,将其应用于Zn-NO3-电池(图4a),因此,采用改进的H型电解槽制备了Zn-NO3-电池,其中碳纸(CP)负载的NSC和Fe1/NSC作为阴极。首先,Fe1/NSC/CP‖Zn电池的开路电压(OCV)约为1.756 V,而NSC/CP‖Zn电池的OCV约为1.547 V(图4b)。Fe1/NSC/CP‖Zn和NSC/CP‖Zn电池的放电极化曲线如图4c所示。随着外加电压的降低,NSC/CP‖Zn和Fe1/NSC/CP‖Zn电池的输出电流逐渐增大。Fe1/NSC/CP‖Zn电池的最大功率密度达到4.42 mW cm-2,是NSC/CP‖Zn电池(2.25 mW cm-2)的近2倍。这些结果表明,Fe1/NSC优异的NO3RR活性有利于实现高输出功率密度。
此外,还研究了不同电流密度下的随时间变化的放电性能。如图4d所示,Fe1/NSC/CP‖Zn电池在0.1、0.2、0.4、0.6、0.8、1、2 mA cm-2的电流密度下表现出良好的电化学稳定性。测定了不同电流密度下的NH3产率和法拉第效率。NH3产率随放电电流的增大而增大,而法拉第效率先增大后减小,这可能是由于在高电流密度下发生的副反应降低了NH3的法拉第效率(图4e)。此外,长期稳定性测试表明,该装置可以连续工作16小时以上,没有明显的衰减(图4f)。上述结果证明了Fe1/NSC/CP‖Zn电池的长期稳定性。
文献信息
A General Strategy Based on Hetero-Charge Coupling Effect for Constructing Single-Atom Sites,Angewandte Chemie International Edition,2024. https://onlinelibrary.wiley.com/doi/abs/10.1002/anie.202408771















 364
364

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









