










 文章详细描述了C3N单层的化学性质,通过第一性原理计算发现其在空气暴露时具有更多的O2吸附位点和强吸附能力,特别探讨了氧分子在C3N上的物理吸附和化学解离过程。研究结果对C3N的催化和材料工程应用有重要指导意义。
文章详细描述了C3N单层的化学性质,通过第一性原理计算发现其在空气暴露时具有更多的O2吸附位点和强吸附能力,特别探讨了氧分子在C3N上的物理吸附和化学解离过程。研究结果对C3N的催化和材料工程应用有重要指导意义。

成果简介
受到石墨烯研究取得巨大成功的影响,各种二维材料相继出现并在电子、催化、气体传感器、能量转换和存储等方面不断发展,同时人们越来越关注其在各种条件下的化学稳定性。特别是,暴露在空气中的二维材料氧化过程中的O2解离是纳米材料操作和器件工程中不可避免的关键问题。最近,一种化学计量公式为C3N的无孔类石墨烯二维蜂窝结构被成功合成,并被证明具有超高刚度和优异的导热性、导电性,在光电催化剂、气体吸附介质和电池电极材料等方面具有潜在的应用前景。单层C3N暴露在空气中的氧解离和氧化结构是C3N工程和表面功能化的关键,但其机理尚未揭示。
扬州大学涂育松等人利用第一性原理计算发现原始单层C3N比原始石墨烯具有更多的O2物理吸附位点和更强的O2吸附能力。
计算方法
本文计算基于VASP模拟包中实现的密度泛函理论进行,采用广义梯度近似(GGA)下的PBE泛函计算电子交换相关性,价电子与离子之间的相互作用由投影增广波(PAW)方法描述。本研究采用grime的DFT-D3方法描述非局域色散相互作用。平面波的能量截止值为450eV,能量和力的收敛准则分别为1×10-5eV和0.02eV/Å。真空层沿z轴方向设置为20Å,以避免周期性结构之间的相互作用。使用Monkhorst-Pack方案对布里渊区的k点网格进行采样,结构优化采用3×3×1的k点网格,对于态密度(DOS)计算,我们使用更大的12×12×1的k点网格来保证精度。所有计算均采用3×3×1的超胞大小,其中包含54个碳原子、18个氮原子和2个氧原子,并且系统完全松弛,直到满足收敛准则为止。采用CI-NEB方法搜索O2分子解离途径。所有的过渡状态(TS)由默认频率计算算法输出的单个虚频率来确定。
结果与讨论
3×3×1的单层C3N超胞如图1所示,包括54个碳原子和18个氮原子。优化后的C3N晶格常数为a = b = 4.86 Å,与实验值一致。
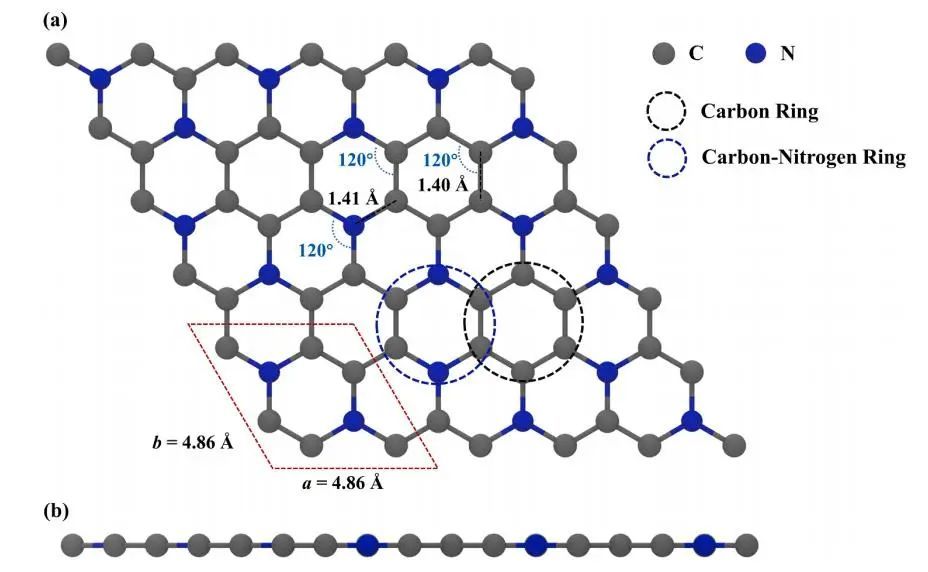
图1 3×3×1的单层C3N示意图
O2分子的物理吸附构型如图2所示,共发现8种稳定的吸附构型,可分为两种类型:六元碳环上的吸附位点(图2a-d))和六元碳氮环上的碳氮环(图2e-j)。
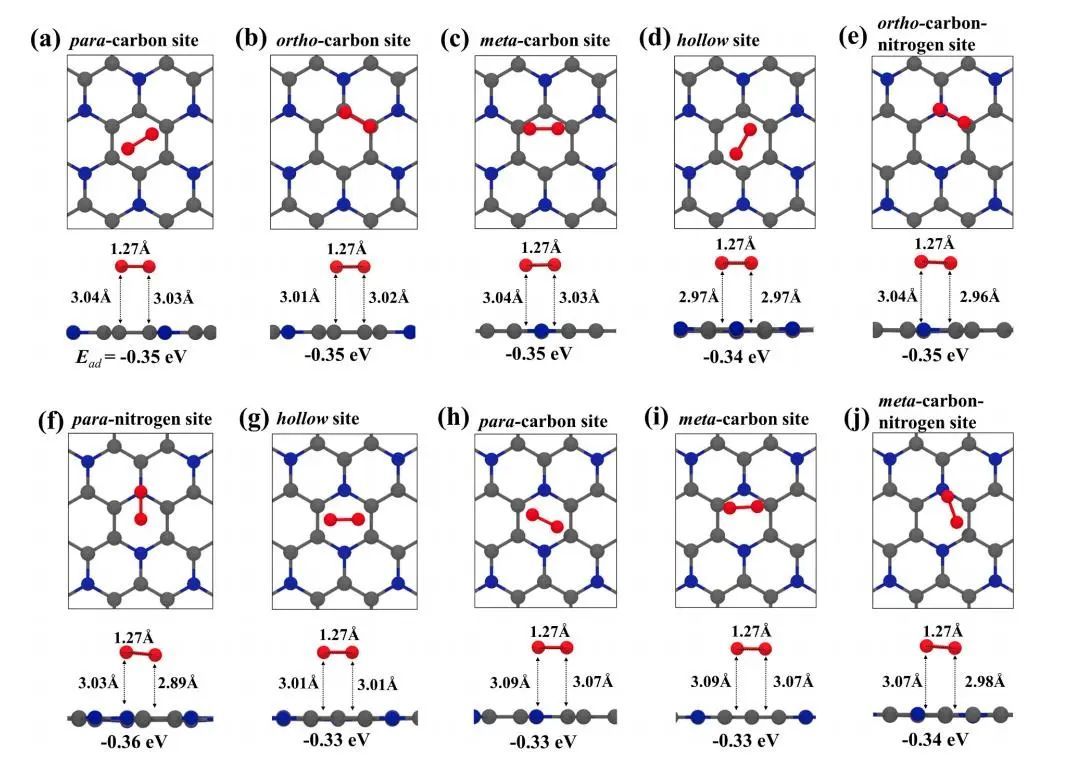
图2 优化后的O2分子在原始单层C3N上的物理吸附构型
与原始石墨烯相比,原始单层C3N显示出更多的O2物理吸附位点,并表现出更强的O2吸附能力。O2分子可以吸附在碳环(对碳、邻碳、间碳和中空)或碳氮环(邻碳氮、对氮、中空、对碳、间碳和间碳氮)上的几个位点上。这些位置的吸附能接近0.34 ~ 0.36 eV,比原始石墨烯的吸附能低2倍。原始单层C3N上O原子的化学吸附位点如图3所示,O2分子解离成两个化学吸附的O原子。作者研究了四种可能的化学吸附位点:碳原子(TC)和氮原子(TN)的顶部位点,C-C键(BC-C)和C-N键(BC-N)的桥位。O原子可以以C-O-C(环氧)的形式停留在BC-C位点,而不能稳定存在于BC-N位点上。对于石墨烯,O原子只能停留在BC-C位点,其吸附能Ead= -2.32 eV高于C3N上的-3.61 eV,表明环氧结构在C3N上更稳定。N原子的存在丰富了C3N表面的各种化学键,如悬垂的C-O和N-O键,这些吸附结构在原始石墨烯上是热力学不稳定的。
 图3
图3
优化后的O原子在原始单层C3N上的化学吸附示意图
氧在碳环上解离:路径I和路径II为解离过程只有一步,反应物(R)和生成物(P)仅通过一个过渡态(TS)连接,如图4所示,解离势垒分别为1.81 eV和1.54 eV。O2在空心位点上的物理吸附构型(图2d)可以通过O2的平移,克服0.62 eV的势垒,转移到中间碳位点上的物理吸附构型(图2c)。路径III是一个间接的两步过程,涉及O2的化学吸附作为解离的中间体,具有相对较低的势垒,为1.10 eV。
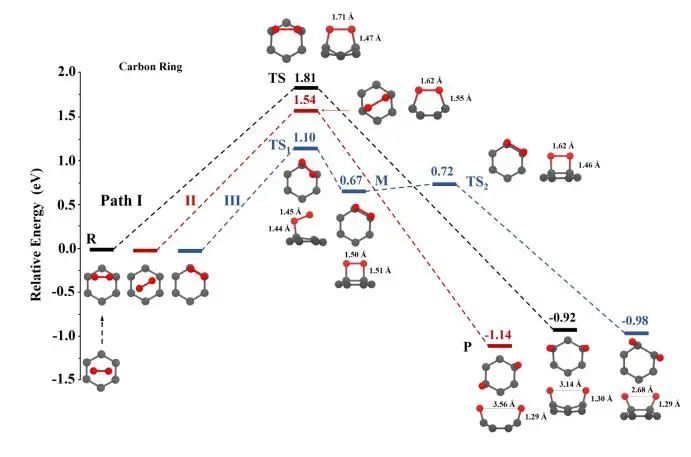
图4 单层C3N碳环上O2解离途径及优化构型
氧在碳氮环上的解离:碳氮环上的路径I-V均为一步解离过程,其势垒分别为4.44 eV、2.86 eV、2.60 eV、1.74 eV和1.34 eV。路径I-III中解离势垒明显比碳环上的大很多,并且通过反应能量变化发现解离是吸热的。路径IV和V中解离是放热的。
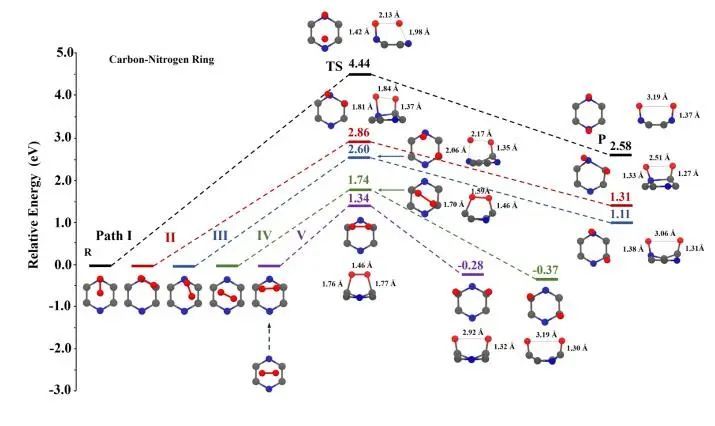
图5 单层C3N碳氮环上O2解离途径及优化构型
原始单层C3N上O2的解离途径取决于初始物理吸附位点和氧化结构,最理想的解离途径是两步过程,其中中间体将化学吸附的O2吸附到碳环的邻碳原子上,解离势垒为1.10 eV,低于原始石墨烯上类似过程的约2 eV。结果表明,原始单层C3N比原始石墨烯更容易氧化。在原始单层C3N中,氮原子的存在有助于形成稳定的悬垂C-O或N-O键,导致氧解离后的富集氧化结构。对于解离态,两个O原子倾向于以悬垂的C-O或N-O键的形式稳定地化学吸附到碳或氮原子上。在碳环的对碳原子上有两个悬垂的C-O键的氧化结构显示出最低的吸附能,为-8.16 eV,比其他结构更有利。
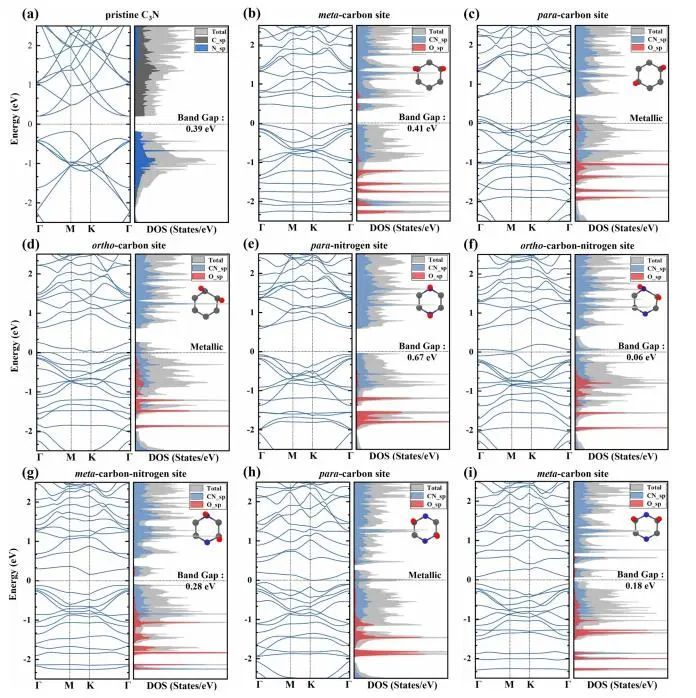
图6 不同构型的能带和态密度
原始单层C3N为间接带隙,带隙为0.39 eV。如图6所示,在碳环的对碳位、邻碳位和碳氮环的对碳位上具有两个O原子的单层C3N的价带穿过费米能级。这表明氧化后的单层C3N由于两个O原子与裸单层C3N在价带边缘的轨道耦合而变成了金属。而在碳环的中间碳位和碳氮环的对氮位上的O原子,轨道杂化主要发生在远离费米能级的低能处,带隙分别扩大到0.41 eV和0.67 eV。O原子在碳氮环的邻碳氮位、间碳氮位和元碳位上的能带宽度分别为0.06 eV、0.28 eV和0.18 eV,表明O原子的电子活性增强。这些结果表明,由于两个O原子的化学吸附,单层C3N的电子性质发生了显著的变化,从金属到半导体,最大带隙为0.67 eV。作者研究了8种可能的氧化结构中O2的吸附吉布斯自由能 ΔGad与O的化学势 ΔμO之比。在一定的ΔμO下,在碳环的对碳位上的两个O原子的化学吸附的ΔGad值低于其他构型的值,表明该构型是最有利的氧化结构。发现即使是碳环的对碳位在自然环境条件下C3N或石墨烯上的自发O2解离依旧非常困难的。
结论展望
作者通过第一性原理计算,系统地研究了氧分子和氧原子在原始单层C3N上的不同吸附位点、8种氧解离途径和相关的氧化结构。为C3N或其他氮化碳衍生物的催化和材料工程研究具有重要意义。
文献信息
Zhao, L., Luo, W., Huang, Z., Yan, Z., Jia, H., Pei, W., & Tu, Y. (2023). Oxygen dissociation on the C3N monolayer: A first-principles study. Applied Surface Science, 613, 155912.
htt-ps://doi.org/10.1016/j.apsusc.2022.155912


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









