










引言
锂离子电池的爆炸式增长电池消费电子产品、电动汽车和储能系统市场已经彻底改变了社会和能源行业。阴离子氧化还原化学通常由蜂窝状有序的富锂层状氧化物中的Li-O-Li构型引发,它已引起越来越多的关注,以从活性晶格氧的主体框架中获得额外的容量。
然而,对于地球上丰富的3d过渡金属(TM)来说,TM和氧(TM-O)之间的共价相互作用不足,不足以稳定主体框架,特别是在深度脱锂状态下,导致氧过度氧化形成氧空穴、O-O二聚体,甚至分子O2。随后,这种不稳定的氧化晶格氧引发了不可逆的TM迁移(面内和面外),导致结构无序、构型重排和主体框架崩溃,同时伴随着电压滞后和电压衰减(图1a)。
这些亟待解决的瓶颈阻碍了富锂层状氧化物的商业应用。实验和理论计算都表明主体框架与TM迁移和氧氧化还原可逆性密切相关。TM迁移通常与氧氧化还原相结合,已被证明是初始和后续循环中电压滞后的根源。此外,通过TMO6单元脱配形成的短氧O-O 二聚体(<1.5 Å)已被证明是结构无序的驱动力,尽管它们也被认为可以稳定氧化氧。这种脱配位是由高脱锂状态的主体骨架不稳定引起的,易于形成短而强的共价键,通过键重排形成稳定的化学态。键重排的趋势与TM-O键的共价相关。
然而,富锂层状氧化物仍然缺乏通用的材料设计规则来实现结构稳定和电化学可逆的氧氧化还原化学。最近提出了一种通过4d 和5d TM 取代增强TM-O共价来稳定氧化氧的替代策略。然而,适当的阳离子取代对于重且贵重的TM离子(Ru、Sn、Ir等)来说是有限的,这不可避免地损害了能量密度并增加了正极材料的成本。除此之外,还有一个异常的样品,其中较大的TM-O共价键可能不利于结构稳定性,因为它容易诱导过多的电子从氧转移,导致形成更不稳定的氧氧化。
另一种可行的方法是调整TM 层的堆叠顺序。与O3 堆叠相比,由于TM 和Li 层的局部结构不同,O2堆叠序列已被证明可以抑制TM迁移。最近,有人提出了第三种方法,涉及用更多共价硫取代氧,这足以保证TM配体稳定性和可逆阴离子氧化还原化学。人们对硫氧化还原化学的兴趣重新燃起,促使研究人员重新探索富锂层状硫化物。
正文部分01成果简介
近日,南京大学的周豪慎教授与郭少华教授等人使用具有强TM-S共价性的硫基主体来缓冲高度活化的硫氧化还原过程中的晶格畸变,并在稳定主体框架方面取得了巨大的成功。实验结果表明,在Li+脱嵌/嵌入过程中,层状硫晶格(尤其是蜂窝状超晶格)得到了长期保存,而富锂氧化物中的结构发生了大幅退化。此外,富锂硫化物正极表现出可忽略不计的0.08 V过电势和0.13 mV/周循环的压降,同时在循环时保持显着的可逆容量。
这些优异的电化学性能可以明确地归因于硫的轨迹比氧的轨迹短得多,这是通过高维神经网络在大尺度(∼30 nm)和长时间尺度(∼300 ps)的分子动力学模拟中揭示的。脱锂过程中的网络电位。我们的研究结果强调了稳定主体框架的重要性,并为设计具有耐用阴离子氧化还原化学的富锂正极建立了一般指导。该研究以题目为“From Oxygen Redox to Sulfur Redox: A Paradigm for Li-Rich Layered Cathodes”的论文发表在国际顶级期刊《Journal of the American Chemical Society》。
02图文导读

【图1】稳定层状富锂主体框架从氧化物转变为共价硫化物的替代策略。
(a) TM和氧周围的配位环境示意图。结构重排通常伴随着阴离子氧化还原过程,导致TM迁移、TM-O键缩短以及脱锂时的氧二聚。
一般来说,在初始状态下,配位环境中典型的TM-O和O-O距离分别>1.9 Å和>2.4 Å。(1,3)在脱锂过程中,在不可逆的氧氧化还原和TM迁移的驱动下,形成了短TM-O键(<1.9 Å)、O-O二聚体(<1.5 Å),甚至分子O2。(1,3)这些相互作用导致键重排,从弱π型相互作用转向更稳定和离域的π型相互作用(短TM-O键形成)和σ型相互作用(O-O二聚化),这通常被认为是TM- O 脱协调过程。(b) TM和配体之间从氧化物到共价硫化物的π型共价相互作用。
一般来说,TM-S键的TM 3d轨道与S 3p轨道的重叠程度高于TM-O键的O2p轨道的重叠程度,这可能归因于硫的大电子轨道。此外,由于极化效应,与TM-O键相比,TM-S键的轨道很容易扭曲,以保持TM-S键的最大重叠。因此,电子分布在TMS6八面体中更加离域,从而产生了强大的π型相互作用系统,使其能够承受一定程度的结构畸变。这种强共价体系有利于循环时主体框架的电荷转移和稳定。
相反,由于TM-O键的π型相互作用较弱,轨道畸变可能导致氧化物中轨道重叠减少,甚至键重排,这不利于结构稳定性。(c)典型富锂层状氧化物(Li2MnO3)和硫化物(Li2TiS3)等值面等值线图中的ELF分布(ELF = 0.2)。在Li2MnO3中,电子主要集中在氧周围,氧和TM原子之间的区域几乎没有电子分布,这表明TM和氧之间具有离子键特性(红色循环区域)。相反,Li2TiS3中ELF的等效球体在TM和硫之间连接并重叠,表明存在很强的共价相互作用。
整体和局部结构特性
LLS和 LLO都具有 O3型层状结构,TM层中具有蜂窝状有序单元,粉末XRD 数据证实了这一点(图2a、图S2 以及表S1 和S2)。特别是,通过Rietveld精修测量的高分辨率同步加速器XRD 图案表明,LLS主体具有更大的晶胞(a = b = 3.5451 Å,c = 17.9351 Å),可以容纳具有大核半径的硫原子。与LLO 类似,LLS中也观察到位于8-10°的几个未索引峰,并被识别为超晶格峰,这表明存在蜂窝有序超结构,即TM层中的Li@TM6单元。
此外,7Li魔角旋转核磁共振(MAS NMR)光谱清楚地显示了宽信号,占Li+的19.96%(浅红色区域),以及具有对称旋转边带(蓝色星号)的更清晰的信号,占Li+的80.04%,这与Li+分别在TM层和Li层占据位点的不同局部环境有关(图2b)。为了更深入地了解原子尺度的晶体结构,采用先进的球差校正扫描透射电子显微镜(STEM)结合高角度环形暗场(HAADF)和环形明场(ABF)成像来直接观察原始LLS 中的原子排列(图2c、d和图 S3)。
HAADF-STEM图像中的亮点对比度和ABF-STEM图像中的暗点对比度揭示了TM和硫的位置,而ABF图像中的亮点对比度对应于Li的轻元素柱。HAADF-STEM和 ABF-STEM观察结果均表明LLS 以O3 型堆叠顺序结晶,这与沿[010]晶体方向观察的原子模型非常匹配。同样,在HAADF-STEM图像中沿[001]区带轴观察到TM离子呈六方对称排列,这也与O3型原子模型一致(图3)。
通过选区电子衍射(SAED)图案进一步证实了TM层中蜂窝状上部结构的直接证据,如图2e所示。尖锐亮点(黄色圆圈)的SAED图案完全索引到R3̅m晶胞,而主要斑点周围的其他相对较暗的反射(红色圆圈)则索引到沿[001]p方向的理想三角P3112上层结构。能量色散X 射线光谱(EDS) 绘图清楚地验证了Ni、Ti和 S均匀分布在所选颗粒中(图2f-i)。

【图 2】蜂窝有序LLS 样品的结构表征。(a) 同步加速器XRD 和Rietveld 细化图案(波长λ = 0.774893 Å),其中包含数据点(红色圆圈)、计算轮廓(黑色圆圈)、差异(蓝线)和布拉格位置(绿线),如图所示。
插图显示了放大的区域,具有明显的蜂窝状有序产生的超晶格峰。(b) 实验7Li MAS NMR 谱和LLS样品的拟合结果。对称旋转边带标有星号。定量分析表明大约19.96%的Li+位于TM层中。(c) 高分辨率HAADF-STEM 图像,显示LLS 样品沿[010]区轴的原子排列,显示重金属(Ni、Ti)和硫原子的对比度。沿[010]观察的原子模型叠加在(c)中,其中浅红色和浅黄色球体分别代表TM原子和硫原子。(d) ABF-STEM 图像显示LLS 正极沿[010] 区轴的TM 原子排列。观察结果与沿[010] 晶体方向观察的O3 型原子模型很好地匹配,如(d) 中插图所示的可视化。(e) 沿[001] 区域轴的SAED图案。黄色圆圈表示中心和基本衍射点,这是R3̅m空间群所共有的。红色圆圈点对应于上层建筑峰值。(f–i) HAADF图像和相应的Ni、Ti和 S元素映射。
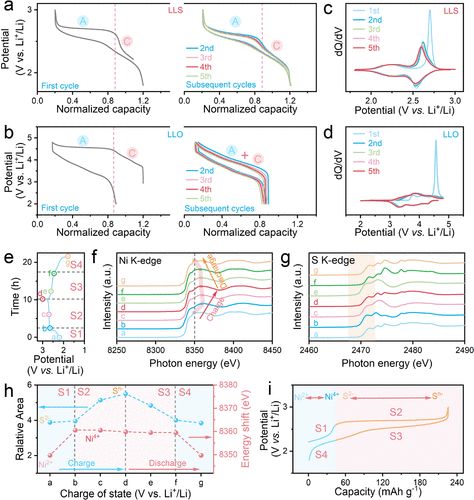
【图3】硫基和氧基主体正极中的电压行为和氧化还原对。
(a)第一个和后续循环在20 mA g–1下获得的LLS 正极电压分布在1.8 和3.0 V之间。
请注意,在充电和放电过程中,后续电压曲线仍然表现出单独的阳离子和阴离子氧化还原平台(分别表示为C 和A)。(b)第一个和后续循环20 mA g–1在2.0和 4.8 V 之间获得的LLO正极电压曲线。
请注意,高压平台(氧氧化还原)在后续循环中消失,表明阳离子氧化还原和氧氧化还原是耦合的。(c, d) d Q /d V曲线源自LLS 和LLO 正极的第一电压曲线。(e) 选择充电和放电状态来研究LLS正极在初始循环中的电荷补偿机制。根据可能的电荷补偿机制,电压曲线分为四个阶段(标记为S1、S2、S3和S4)。具体点在电压曲线中进行了标记。(f, g)各种充电和放电状态下的非原位Ni 和S K 边缘XANES 光谱。(h) SK 边缘约2472 eV处的前边缘峰值的相对面积的演变以及初始循环期间Ni K 边缘的能量偏移(白线峰值)。
Ni K 边缘的白线峰值在阶段S1 中转移到更高的能量,并在阶段S4中返回到其初始值。与之互补的是,前边缘峰(S K-edge)的积分面积在S2阶段明显增加,而在S3阶段明显减少。(i)初始充放电过程中的累积电荷补偿。
硫基和氧基富锂层状正极的电压行为
电压分布可以提供有关充电和放电过程中电子和结构变化的重要信息。图3a、b展示了 LLS和 LLO电极在初始和后续循环期间电压演变的比较。在第一个循环的充电过程中,两条电压迹线均表现出富锂层状正极的典型形状。具体来说,早期的短斜电压特征主要对应于TM氧化还原活性(标记为C阶段)。随后,在进一步氧化之后,出现一个可能与阴离子氧化相关的长平台(标记为阶段A)。也就是说,电压曲线中LLS 的阶段C 和阶段A 最有可能分别与阳离子(Ni2+至Ni4+)和阴离子(S2–至S(2–n)–,n<0.5)氧化有关。
同样,正如开创性工作所确定的,LLO正极的短倾斜区域(阶段C)和长平台区域(阶段A)最有可能分别与镍和氧的氧化过程相关。然而,在初始放电过程和后续循环期间电压分布的形状有很大不同。对于LLO,初始放电过程的形状发生了变化,并且轮廓移动到比充电过程低得多的电势方向,导致高达1000 mV的大电压滞后。如此大的电压滞后(第一周期)可以认为是由伴随TM迁移的部分不可逆氧氧化还原以及蜂窝状上部结构的损失引起的。
相比之下,LLS的电压分布在放电过程的早期阶段仍保持较长的平台期,随后出现短斜率电压特征,导致电压迟滞可忽略不计,仅为 ∼0.08 V。这种对称性也在随后的循环表明LLS可能经历不寻常的对称氧化还原途径,这与LLO有本质上的不同。相应的微分容量图也初步证实了LLS的对称氧化还原路径(图3c)。在充电和放电过程中,两个氧化峰保持在2.2和2.6 V,分别对应于阳离子(Ni2+)和阴离子(S2–)氧化过程。
请注意,S2–的氧化峰比Ni2+的氧化峰强得多,说明阴离子氧化还原贡献主导了LLS正极的容量。值得注意的是,后续循环(第二至第五次)的微分容量曲线完全重叠,进一步证实了阳离子和阴离子氧化还原的高度可逆过程。为了进行比较,在LLO中,高电位阴离子氧化还原的平台在后续循环中消失,表明阴离子氧化还原与阳离子氧化还原耦合(图3b、d)。这种现象与在典型富锂层状氧化物正极中观察到的大电压滞后现象非常吻合,这种现象源于氧氧化还原和TM迁移之间的强耦合。
阳离子和阴离子氧化还原的对称电荷补偿机制
最近在大多数富锂层状氧化物正极材料中观察到阴离子氧化还原和阳离子迁移之间的内在耦合机制。人们普遍认为,TM从TM层迁移到Li层会降低氧的氧化还原电位,导致循环时氧化还原化学不对称。这种不对称反应路径被认为在激活电压迟滞以及循环过程中的电压衰减中发挥着重要作用。在这里,考虑到循环过程中的对称和可逆电压分布,预计LLS的氧化还原路径不同于传统的富锂层状氧化物。对于原始样品,确认Ni的初始价态为Ni2+(图S4)。此外,先前的研究表明,由于空的3d轨道构型,硫化物中的Ti4+不活跃。在具有明显充电和放电状态的初始循环期间,通过异位XANES仔细研究了氧化还原电对的演变(图3 e-i 和图S5)。
原则上,Ni和Ti K边的能量偏移可以从白线峰的位置确定,而S的价态可以从前边峰的相对面积间接确定,这已被被认为是确定TM和硫价态的可靠方法。在充电和放电状态下,Ti K 边几乎没有能量变化,进一步表明Ti 是结构框架的非活性成分(图S5)。因此,只有Ni和S参与LLS中的电荷补偿。如图3f所示,在充电过程的倾斜区域(从a到b,标记为S1),Ni K边缘的白线峰向更高光子能量移动,而在平台期间几乎保持不变即使达到完全充电状态(从b到d,标记为S2),该区域仍然存在,表明仅在阶段S1中检测到Ni氧化还原(Ni2+到Ni4+)。
同时,SK边缘的前边缘峰面积在S1阶段几乎保持不变,在S2阶段明显增加,表明S在较高的脱锂状态下明显失去电子(图3g)。这些结果表明,Ni和S参与了对应于两个独立充电阶段的电荷补偿,这与电压曲线和微分容量图一致(图3a,c)。值得注意的是,放电过程中Ni 和S K边缘的变化与充电过程中的变化是对称的(图3 e-h)。也就是说,在放电过程结束时,白线峰(Ni K-边缘)的位置和S前边缘峰的相对面积都完全恢复到其初始状态,这意味着高度可逆和对称的阳离子和阴离子氧化还原化学是在LLS 中实现的(图3 h)。
结合上述结果,我们在图3i中总结出阳离子和阴离子氧化还原化学遵循对称途径。具体而言,阳离子(Ni2+至Ni4+)氧化还原仅出现在较低电荷状态,而阴离子(S2–至S(2–n)–,n < 0.5)氧化还原在较高脱锂状态下单独参与电荷补偿状态。
循环时蜂窝状上部结构的保留和结构演变
最近报道的许多工作表明,电压分布与涉及TM迁移的晶体结构的演化密切相关。TM迁移,包括面内迁移和面外迁移,被提出作为配置重排的关键证据,以解释初始周期中的电压滞后和后续周期中的电压衰减。对称的电压分布和可忽略的电压滞后(~0.08 V)促使我们提出一个假设,即TM 迁移,尤其是面内迁移,在LLS中受到抑制。
基于这一假设,进行非原位XRD 来表征循环过程中LLS 和LLO 正极的结构演变,如图4a -d所示。在完全充电状态、完全放电状态甚至经过50次循环后都观察到了保持良好的O3型结构,具有明显的超晶格峰,这表明LLS具有高度稳定的结构,没有面内或面外TM迁移。也就是说,晶格硫的主体框架通过强TM配体共价键得以稳定。特别是,Rietveld精修同步加速器XRD图案和完全充电状态的相应电池参数如图4e和表S3所示。
Ni和Ti K边扩展X射线吸收精细结构(EXAFS)光谱的径向分布函数(RDF)也表明TM离子的局部环境在循环过程中几乎保持不变(图S6)。相比之下,LLO的超晶格峰在初始循环的高脱锂状态中消失,并且在随后的循环中没有重新出现,表明LLO在脱锂过程中经历了不可逆的结构重排。因此,LLO和 LLS正极之间初始电压分布的本质差异与超晶格峰值在循环过程中是否保留密切相关。

【图4】主体框架对超晶格的依赖性以及循环时的结构稳定性。
(a) LLS正极在初始状态、完全充电状态、完全放电状态和50 个循环后获得的异位同步加速器XRD 图案(波长λ = 0.774893 Å)。请注意,蓝色阴影区域中的超晶格峰在整个电化学循环过程中保持不变。(b) (a)中的放大区域,具有明显的蜂窝状有序超晶格峰。(c)在初始状态、完全充电状态、完全放电状态和50 个循环后获得的LLO 正极的异位XRD图案(波长 λ Cu = 1.5418 Å)。(d) (c)中的放大区域表明蓝色阴影区域中的超晶格峰在完全充电状态后消失并重新出现。(e) 完全充电的LLS 样品的高分辨率同步加速器XRD 和Rietveld 细化图案(波长λ = 0.774893 Å),其中包含数据点(黑色圆圈)、计算轮廓(红色圆圈)和布拉格位置(绿线),如表明的。(f) 原位XRD 轮廓图显示LLS正极两个主峰的演变,并提取电池参数的变化以及20 mA g-1电压范围1.8-3.0 V初始循环的相应时间–电压曲线。
为了进一步了解LLS在循环过程中的结构演变,进行了原位XRD来监测前两个循环中的晶体结构变化(图S7)。特别是,初始周期的时间–电压曲线和两个主要峰值(强度等值线图样式)的演变如图4f所示。(003)和(104)峰在充电过程中平滑地向更高程度移动,然后在放电过程中逐渐返回到初始位置,表现出高度可逆的对称固溶体行为。
通过定量分析提取LLS正极的电池参数,以显示它们在初始循环中的变化(图4f)。参数a、参数c和电池体积的变化分别仅为约0.42%、0.99%和1.94%。这种显着的结构稳定性可能归因于强的TM-配体共价相互作用,它可以防止循环过程中ab平面的收缩和c轴的膨胀。因此,LLS正极有望实现优异的电化学性能,特别是快速动力学阴离子氧化还原化学性能。
硫基和氧基主体正极的实际评估
由于LLS在循环过程中高度稳定的结构通过非原位同步加速器XRD和原位XRD得到验证,因此进行了深入研究以突出源自阳离子和阴离子氧化还原化学的优异电化学性能。在电流密度为20 mA g–1时,可逆比容量达到251 mAh g–1(611.1 Wh kg–1) ,与LLO正极相当(图S8)。
此外,LLS在循环时表现出可忽略不计的容量损失(在100 mA g–1下测试),而LLO正极则遭受严重的容量衰减和电压衰减(图5 a、b)。这些结果与LLS 和LLO 的结构演变非常一致(图4 a-d)。此外,请注意,即使在50 个循环后,LLS的电荷平台(~2.6 V)仍然保留,进一步证明基于硫的主体框架抑制了TM 迁移。
为了评估LLS 和LLO正极的反应动力学,我们在第一个循环中进行了恒电流间歇滴定技术(GITT) 测试(图S9)。与LLO 正极相比,LLS的 GITT曲线在整个充电和放电过程中显示出可忽略不计的反应过电势,表明硫氧化还原的动力学比氧氧化还原的动力学快得多。图5c显示了LLS优异的速率性能。它仍然可以在2000 mA g–1的电流密度下呈现180 mAh g–1的大容量和同时高库仑效率。
同时,在2000 mA g–1循环后,当倍率恢复到20 mA g–1时,放电容量可以恢复到初始值,表明LLS正极具有良好的反应动力学和优异的电化学稳定性。然后,测试了高倍率(1000 mA g–1)下的长期循环性能,如图5d所示。即使循环1000次后,LLS正极仍保留161 mAh g–1的可逆容量(80%容量保持率),明显优于LLO正极。此外,LLS正极在100 mA g–1下也表现出~581.5 Wh kg–1的能量密度,并且在50次循环后几乎没有能量衰减,而在LLO正极中观察到快速的能量损失(图S10)。
即使在1000 mA g–1的高电流密度下循环,LLS仍然在 1000次循环中实现了卓越的长期能量保持(图S10)。此外,LLS在1000个循环后的平均电压降仅为~130 mV,而LLO正极在前10个循环中表现出快速电压降,在500个循环后超过1100 mV,如图5e所示。此外,将电压降与最近报道的富锂层状正极材料的电压降进行比较后,LLS正极位于图5f的右角,表明本征电压衰减已被有效抑制。为了进一步评估LLS 和LLO正极的电化学性能,还使用不同的电流密度测试了>10 mg cm–2的更高质量负载。
实验结果表明,尽管初始容量略有下降,但与LLO相比,高质量负载LLS仍然可以表现出优异的倍率性能和循环稳定性(图S11)。此外,整体阻抗的演变表明,LLS正极的电荷转移电阻在长期循环过程中保持较小且几乎恒定的值,而LLO在1000次循环后急剧增加(图S12)。Ni 2p和Ti 2p的XPS结果也表明,在长期循环后,Ni和Ti的价态仍然保持+2和+4(图S13和图S14)。这进一步证明TM和硫之间的高共价性可以强烈稳定TM离子(Ni和Ti),确保循环时连续的硫氧化还原反应。这些结果再次强调LLS 表现出卓越的长期循环稳定性。

【图 5】LLS和 LLO正极的电化学性能。(a, b) LLS 和LLO 正极在100 mA g–1特定电流密度下的电压–容量演变。LLS 正极即使在50 个循环后,电压曲线也完全重叠,而LLO正极则表现出严重的容量损失和电压衰减。(c) 使用20 至2000 mA g–1的电流密度测试LLS 的倍率性能。(d) 在特定电流密度为1000 mA g–1时获得的LLS 和LLO 的循环性能比较。(e) 在特定电流密度1000 mA g–1下测试的长期循环期间LLS 和LLO的电压降比较。电压降定义为每次循环的循环平均放电电压减去初始平均放电电压,平均放电电压由能量除以容量得到。(f)与最近报道的富锂层状正极材料相比的电压衰减。
通过MD 计算关联LLS 和LLO 主机的结构稳定性
可以采用MD模拟来更好地理解脱锂过程中原子尺度的结构演化。为此,从头算分子动力学(AIMD)模拟已被广泛用于探索插层材料的结构变形和扩散特性。然而,AIMD可实现的时间尺度通常仅限于皮秒范围,这使得在充电和放电过程中准确捕获感兴趣对象的动态特性变得困难。
因此,对于多阳离子体系,由于其长程无序特性,必须考虑更大的晶体体积进行MD模拟。此外,为了在不牺牲准确性的情况下增加AIMD模拟的时空尺度,我们的计算中引入了高维神经网络势(HDNNP),其中数据是通过基于力场的MD 运行获得的。特别是,力场是根据DFT数据通过机器学习生成的。同样,采用主动学习方案来确保机器学习生成的力场的准确性(详细信息请参见支持信息注释2)。按照极小值原则保证了所得力场的精度,与DFT结果接近。
因此,力场法可以在更大的时空尺度上进行模拟,克服了DFT方法在尺度上的限制。在此HDNNP-MD方法的基础上,我们首先执行MD方法来研究脱锂过程中硫基主骨架(LLS)和氧基主骨架(LLO)的结构稳定性。当从模拟盒中提取Li+时,在硫基和氧基主体框架中都观察到局部结构变化(图S15)。此外,在透视图中,即使在硫基主体框架中300 ps后,“晶格条纹”(黄线)仍然清晰可见,而氧基主体框架的晶体结构随着Li+的去除而明显塌陷。(图6a)。
此外,基于结构描述符的畸变指数也表明硫基主体框架的畸变指数远小于氧基主体框架,进一步验证了硫基主体实现了优异的结构稳定性框架(图S16)。还根据MD模拟数据计算了基于元素的扩散系数,如图6b所示。与氧基骨架相比,硫基骨架的扩散系数仅为氧基骨架的一半左右,表明硫基骨架在脱锂过程中保持了更稳定的结构。值得注意的是,晶格氧和硫在主体骨架中所占比例最大;因此,结构畸变主要归因于晶格氧和硫的迁移率。通过脱锂过程中晶格氧和硫的位移进一步验证了这种推理(图S17)。
显然,在模拟过程中硫的运动轨迹比氧的运动轨迹短得多。此外,在0-300 ps的变化时间内测量了氧和硫的平均位移,如图6c所示。硫的较小位移(∼1.9 Å)表明晶体结构中的扩散行为可以忽略不计,并且硫仅在其原始位置周围振动。相反,O的较大位移(累积超过3.1 Å)可能导致O迁移的活性区域更大,并带来局部结构变形,甚至O2释放到氧基主体框架中。因此,MD模拟结果表明,在富锂层状硫化物中实现了高度稳定的主体框架,这可以为阴离子氧化还原反应提供有利的条件。

【图6】脱锂过程中硫基和氧基正极主体结构稳定性的机器学习辅助MD 模拟。(a)MD模拟框(2D透视图)以及几种脱锂状态下的氧基主体框架(LLO)和硫基主体框架(LLS)的相应快照。除Li外,所有原子均处于透明模式。即使在硫基主体框架中300 ps后,“晶格条纹”(橙色线)仍然很明显。(b)计算脱锂过程中硫基主体框架和氧基主体框架中阳离子和阴离子的扩散系数值。(c) S 和O 相对于初始位置的均方位移(MSD) 值作为时间(300 ps)的函数。阴影区域面积是平均位移标准差内的范围。
在目前的富锂层状氧化物中,阴离子氧化还原化学通常被认为总是与氧损失、不可逆的TM迁移以及主体框架内的逐渐结构无序有关,这是电化学不可逆性的主要原因。假设对主体框架的损害源于键和结构重排以形成由过氧化的氧驱动的短共价键,这可能是合理的。也就是说,初始的TM-O键太弱,无法稳定过度氧化的晶格氧和高价TM 离子。然而,TM-O共价、氧损失和TM 迁移之间的关系迄今为止尚不清楚。TM-O键足够的共价性已被证实能够增强循环中的氧基主体框架,从而产生高度可逆的氧氧化还原化学。值得注意的是,对比模型是具有强共价键的Li2RuO3,它仍然显示出氧释放的直接证据。
实际上,即使对于更多的4d和5d基富锂层状氧化物,TM-O键的共价仍然不足以抑制键和构型重排。这可能导致人们普遍认为,通过用更多共价的硫取代氧可以进一步增强TM配体共价性。Li2FeS2、Li1.33–2y/3Ti0.67–y/3FeyS2和NaCr1–yVyS2循环过程中硫基层状结构框架支持了这一点。与氧基主体框架中TM-O键很容易断裂形成过氧化的O-O二聚体或O2不同,强共价TM-S键即使在高电荷状态下也能承受一定程度的结构变形。也就是说,TM-S键可以为氧化硫提供稳定的主体框架,并在长循环过程中表现出持久的蜂窝状有序结构(图4a,b)。
相反,氧基框架不能稳定氧化氧,并且在循环时表现出明显的结构紊乱(图4c,d)。一些例子,例如Li1.2Ni0.13Co0.13Mn0.54O2、Li2–xIr1–ySnyO3和Li2Ru1–ySnyO3,在充电状态下也会失去蜂窝状有序性,并且表现出明显的O2释放和TM迁移,导致初始循环中严重的电压滞后和电压衰减。
此外,我们的探索还可以为一类扩展的阴离子基材料中阴离子氧化还原化学的电荷转移动力学提供有价值的见解。值得注意的是,S (3p) 到 TM (3d) 电荷转移的速率比O (2p) 到TM (3d)电荷转移的速率要快得多,这由阴离子中较小的电压滞后表明。氧化还原过程(图3a -d)。这也与充放电过程中对称的阳离子和阴离子氧化还原途径一致(图3 e-i)。可以解释如下:共价越高,TM与配体之间的极化性越大,离域效应越强,导致电荷转移的动力学势垒较低,从而降低阴离子氧化还原化学的电压滞后。在其他化合物中也观察到类似的趋势,从氧化物到硫化物甚至硒化物。
总结和展望
综上所述,通过掺入更多共价硫有助于在富锂层状正极材料中构建稳定的层状主体框架,并且在LLS正极中实现了硫氧化还原化学的高活性和可逆性。此外,LLS表现出约 0.08 V 的低电压迟滞,并在1000次循环后实现了接近于零的电压降(每循环0.13 mV),优于大多数富锂层状氧化物。我们相信,这种范式不仅提供了设计和探索阴离子正极材料的新方向,而且还提供了解决源自固有阴离子氧化还原化学的重大障碍的新途径。
参考文献
Jing-Chang Li, Jiayi Tang, Jiaming Tian, Chen Cheng, Yuxin Liao, Bingwen Hu, Tao Yu, Haoyu Li, Zhaoguo Liu, Yuan Rao, Yu Deng, Liang Zhang, Xiaoyu Zhang, Shaohua Guo, and Haoshen Zhou
Journal of the American Chemical Society Article ASAP
DOI: 10.1021/jacs.3c11569
















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









