








 本文详细介绍了如何在药效团模型中筛选药物,步骤包括选择配体来源、调整模型匹配度、配置筛选条件等,适合药物研发人员了解模型应用技巧。
本文详细介绍了如何在药效团模型中筛选药物,步骤包括选择配体来源、调整模型匹配度、配置筛选条件等,适合药物研发人员了解模型应用技巧。
咱们文接上篇,用上次构建好的药效团模型筛选药物:
如上图所示,左击右端“H图标”,出现
add to database creen, 鼠标左击,然后会出现下面的对话框:
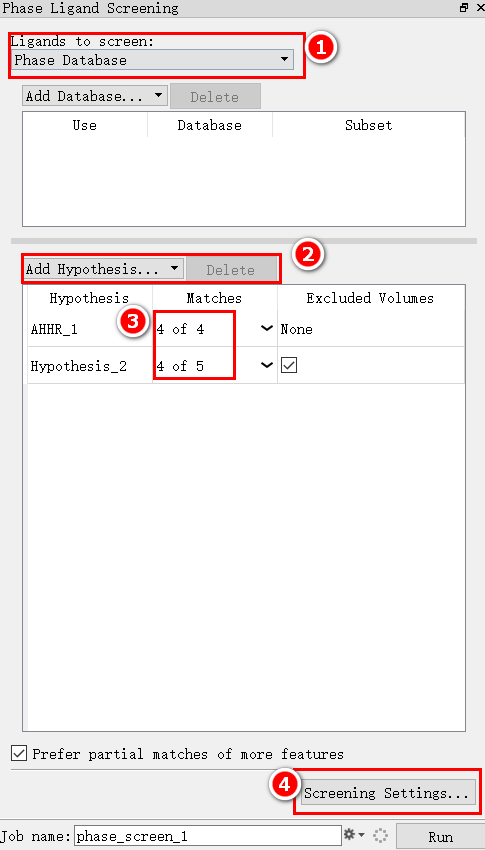
对上图各块内容解读:
- ① 选择配体分子来源:可以是来自导入软件workplace中的分子也可以是直接从本地文件直接读入。如果选择了Phase database,则需要在下方的框内点击add database,在这里添加要筛选的配体。
- ② 可以选中不需要的药效团模型,删除之。
- ③ 此栏信息规定了小分子和模型的匹配度。如可以改为3 of 4,意思是此药效团总共有4个特征元素,筛选的配体分子只要满足其中三个元素就可以被保留下来。
- ④ 关于筛选过程更多设置。


















 3178
3178

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











