











成果简介
对双金属催化剂协同效应的基本认识在多相催化中具有极其重要的意义,但是如何精确构建均匀的双金属位点还面临着巨大挑战。基于此,西北工业大学覃勇教授和任煜京副教授、德国莱布尼茨催化研究所Haifeng Qi、电子科技大学Na Yang等人报道了一种高效的双-单原子催化剂(dual-single-atom catalyst)的构建方法,即通过在纳米金刚石(nanodiamond, ND)表面修饰Fe1-N4单原子位点来锚定Pt单原子。
通过像差校正高-角环形暗-场扫描透射电子显微镜(AC-HAADF-STEM)图像分析、X射线吸收精细结构(XAFS)光谱模拟和X射线光电子能谱(XPS)光谱拟合,证实了纳米金刚石表面存在Pt1-Fe1双-单原子位点(记为Pt1-Fe1/ND)。
测试发现,利用这种Pt1-Fe1双位点,在原子水平上揭示了硝基芳烃化学选择性加氢的协同催化作用。结合实验结果和密度泛函理论(DFT)计算表明,氢在Pt1-Fe1位点被活化,硝基优先在Fe1位点被吸附和活化。虽然Pt1-Fe1位点对Pt NP对应物的氢活化能力稍差,但硝基在Fe1位点上的垂直吸附构型和较强的吸附能使得Pt1-Fe1位点更容易选择性活化硝基。
在Pt1-Fe1双位点上的协同作用使得硝基芳烃加氢具有创纪录的催化性能,其中周转频率(TOF)为3.1 s-1,选择性接近100%,底物适应性达到24种。该发现不仅促进了双-单原子催化剂在选择性加氢中的应用,而且为在原子水平上探索协同催化的本质开辟了新途径。

研究背景
负载型金属催化剂(SMCs)在化工、能源转化和环境保护等领域发挥着重要作用。目前,研究SMCs的协同催化已成为多相催化领域的热点之一。虽然贵金属和非贵金属氧化物间的协同作用在C-O的氢解中取得了很大的进展,但由于传统SMCs中不可避免地存在多种金属位点共存,揭示协同催化的本质仍然是难克服的障碍。
对比传统SMCs,单原子催化剂中金属物种的几何隔离消除了各种活性位点结构(如阶地位点、角位点和阶梯位点)的影响。通过进一步选择性地将另一种金属单原子锚定在单原子催化剂上,以精确地构建异-核M1-M’1双单原子位点,科研人员能够在原子水平上揭示协同催化的基本作用。然而,“自下而上”的合成策略对于构建异-核M1-M’1双位点的可操作性有限。
图文导读
制备与表征
作者首次通过使用Fe1-N4单原子位点(Feδ+)修饰纳米金刚石(ND)表面来锚定Pt单原子前体(PtCl62-),用于自限制表面反应。将酞菁铁(Fe-Pc)前驱体浸渍在ND表面,然后在400 ℃的Ar中处理,在ND表面负载了Fe1-N4单原子位点。接着,通过简单的静电吸附法将Pt前驱体(PtCl62-)负载在Fe1/ND表面,在250 ℃下还原。电感耦合等离子体(ICP)测量显示,Pt1-Fe1/ND样品(2.01 wt.% Pt和1.00 wt.% Fe)和PtNP/ND样品(1.99 wt.% Pt)的Pt和Fe负载量接近理论值。

图1. Pt1-Fe1/ND催化剂的制备示意图和HAADF-STEM图像
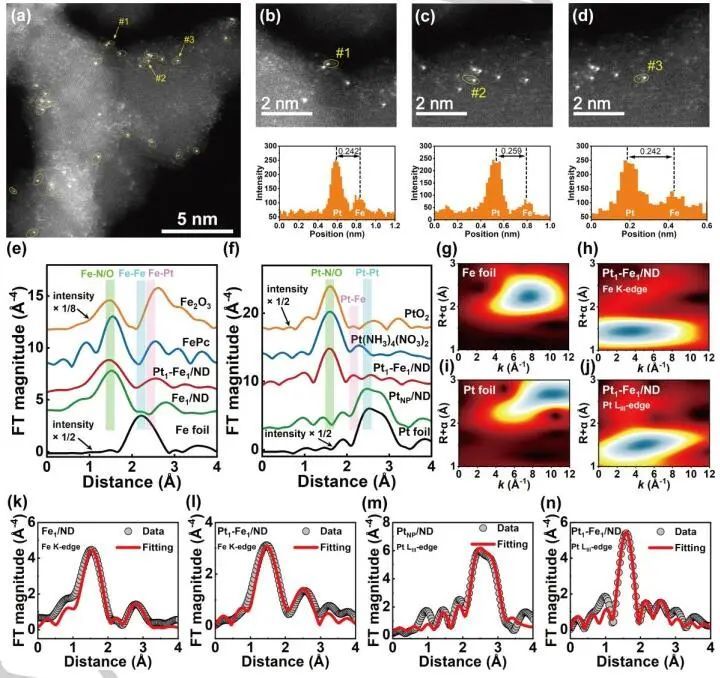
图2. Pt1-Fe1/ND、Fe1/ND和PtNP/ND的AC-HAADF-STEM图像和配位环境表征ATR-IR表征
衰减全反射红外(ATR-IR)光谱采用Thermo Scientific Nicolet iS50光谱仪,配备4 cm-1分辨率的汞镉碲化(MCT)探测器。实验前,将样品分散在乙醇中,超声处理1 h,然后在65 ℃下将悬浮液滴入仪器上的金刚石晶体表面,在100 ℃下干燥4 h。采集数据前,记录背景光谱。
然后,为得到吸附底物的信号,将底物滴加到催化剂表面。H2微热量测量由BT2.15热通量量热计进行,该量热计连接到气体处理和体积系统,采用MKS Baratron电容压力计进行精密压力测量。经计算,微热系统的极限动态真空为10-7 Torr。

图3. Pt1-Fe1/ND、Fe1/ND和PtNP/ND样品的电子结构

图4. Pt1-Fe1/ND和PtNP/ND催化剂上p-NB加氢动力学实验AIMD模拟
通过从头算分子动力学(AIMD)模拟,作者从理论上研究了Pt1-Fe1双单原子位的稳定性。利用密度泛函理论(DFT)计算优化了石墨碳表面N3-Pt1-Fe1-N3的构型,该构型由HAADF-STEM和EXAFS结果推导而来。在353 K条件下,在10 ps内对N3-Pt1-Fe1-N3模型进行了系统的AIMD模拟,以阐明其动态稳定性。
结果表明,在整个模拟过程中,N3-Pt1-Fe1-N3的结构没有发生结构破坏,系统的能量和温度保持稳定,与稳定性实验结果一致。更重要的是,从AIMD模拟的径向分布函数(RDF)结果发现,N3-Pt1-Fe1-N3结构中的Fe和Pt原子分别与键长为1.96 Å和2.01 Å的3个N原子直接配位,并且在2.32 Å处存在明显的Pt-Fe配位,证明了理论上N3-Fe-Pt1和N3-Pt-Fe1在Fe1和Pt1单原子位点上的配位环境。
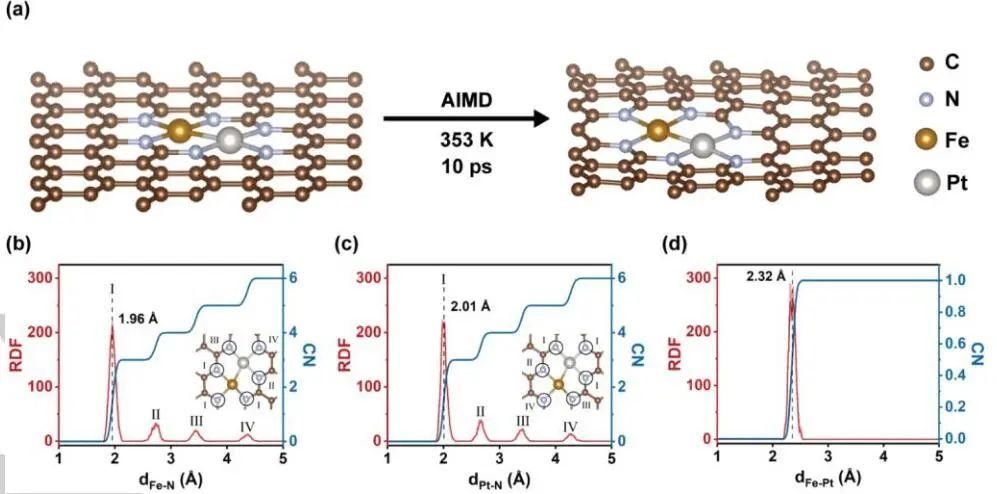
图5. AIMD模拟理论计算
H2在Pt1-Fe1上的吸附和活化,H2分子在Pt1-Fe1双位点上解离,其解离反应势垒(0.50 eV)略高于Pt(111)表面的解离反应势垒,表明Pt1-Fe1较难激活H2分子。p-NB在Pt(111)表面的优化吸附结构,包括平面构型和垂直构型,表明p-NB倾向于在平面结构的Pt表面吸附。对于N3-Pt1-Fe1-N3结构,Pt1-Fe1中的Fe1位点优先吸附-NO2基团在垂直构型的p-NB分子中。对于双位点,Fe1-Fe1和Pt1-Pt1位点均能解离H2,但p-NB分子对Fe1-Fe1的吸附太强,对Pt1-Pt1的吸附太弱。
结果表明,Fe1、Pt1、Fe1-Fe1和Pt1-Pt1位点不利于p-NB的选择性加氢。因此,Pt1-Fe1/ND双-单原子催化剂对硝基芳烃选择性加氢反应表现出优异的催化性能。对于N3-Pt1-Fe1-N3结构,在p-NB加氢反应中存在14个中间态(IMs)和6个过渡态(TSs),发现其表面比Pt(111)表面具有更强的硝基加氢活性。硝基在Fe1上的垂直吸附构型和较强的吸附能,使得N3-Pt1-Fe1-N3更容易活化硝基,导致了优异的催化性能和优异的底物适应性。
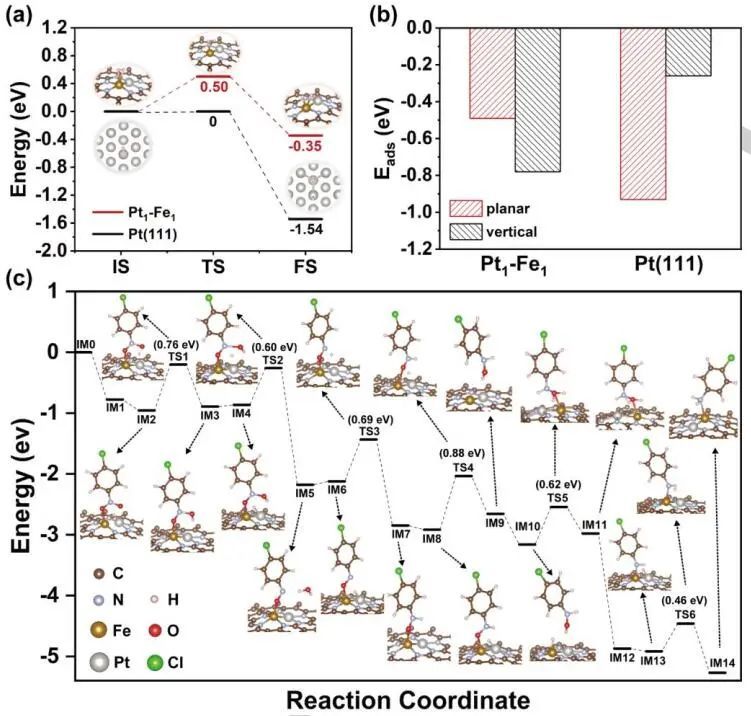
图6.催化加氢过程的机理
文献信息
Atomic Insights into Synergistic Nitroarene Hydrogenation over Nanodiamond-Supported Pt1-Fe1 Dual-Single-Atom Catalyst. Angew. Chem. Int. Ed., 2023















 194
194











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









