










 研究人员通过DFT计算,评估了Co、Fe、Ni、Mn和Mo掺杂钒碳化物(VC)在不同晶面的氢进化反应(HER)性能。研究发现,掺杂后尤其是Mn和Co在(100)和(110)晶面上显示出高效HER,且掺杂能调控电子结构,优化H吸附和解吸过程。
研究人员通过DFT计算,评估了Co、Fe、Ni、Mn和Mo掺杂钒碳化物(VC)在不同晶面的氢进化反应(HER)性能。研究发现,掺杂后尤其是Mn和Co在(100)和(110)晶面上显示出高效HER,且掺杂能调控电子结构,优化H吸附和解吸过程。

研究背景
高效低成本的析氢电催化剂的合理设计是未来能源的重要举措。在本研究中,北京邮电大学符秀丽等人评价了过渡金属(Co、Fe、Ni、Mn和Mo)掺杂钒碳化物(VC)的析氢反应(HER)性能。此外,在(100)、(110)和(111)晶面上分别对掺杂原子进行了筛选,分析了HER活性的差异。
计算方法
本文采用vasp软件包对其催化性能及电子性质进行理论研究,计算基于广义梯度近似(GGA)下的PBE泛函计算电子交换相关性,离子-电子相互作用采用PAW方法进行表征。所有计算都考虑自旋极化。Z方向真空度设置为15 Å,截断能设置为520 eV,将最大力的收敛公差设为0.05 eV·Å-1,能量收敛公差在0.0001 eV。
结果与讨论
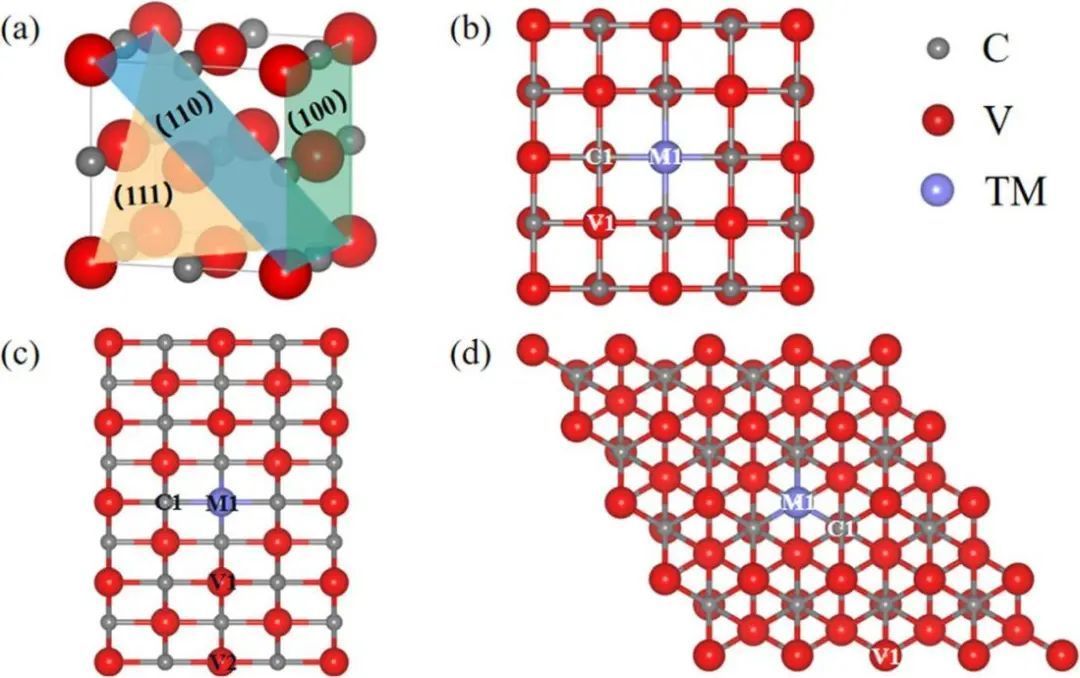
图1 催化剂结构示意图 图1(a)为催化剂不同切面方向的示意图。图 1(b)~(d)为不同表面下进行过渡金属掺杂的示意图。
在比较所构建模型的析氢性能之前,考虑了上述模型的稳定性,以估计TM掺杂后的特定晶面是否能稳定存在。本文对TM掺杂剂的结合能(Eb)和内聚能(Ecoh)进行了评价,研究了掺杂前后VC不同表面掺杂催化剂模型的稳定性,如图2所示。一般来说,如果Eb小于Ecoh,则认为掺杂的TM原子更倾向于插入衬底中,而不是凝聚在一起。图2(a)为VC(100)晶面上掺杂的不同金属的能量变化值,其中黑线表示Ecoh,红线表示Eb。在掺杂不同TM原子的情况下,所有掺杂模型的结合能都小于相应金属的内聚能,说明掺杂TM的VC(100)具有较高的可行性。
与其他掺杂模型相比,Mo-VC(100)的结合能最低(11.05 eV),而Mo的Ecoh为6.25 eV,说明Mo-VC(100)具有显著的相对稳定性。考虑到图2(b)中TM掺杂VC(110)和图2(c)中TM掺杂VC(111)的情况,发现所有计算得到的模型的Ecoh值小于相应金属的Ecoh值,表明TM-VC模型具有稳定性。这些TM掺杂VC模型的三个晶面上的低结合能保证了这些含有掺杂原子的表面的稳定存在。
众所周知,Eb减去Ecoh的值等于ΔEb,在本文中,ΔEb可用作表面模型稳定性的描述符该值越负,对应的曲面模型越稳定。如图2(d)所示,TM原子在(100)平面上的掺杂比在(110)和(111)平面上的掺杂释放出更多的能量,说明TM原子在(100)平面上容易掺杂,且TM掺杂VC(100)模型总体上更加稳定。
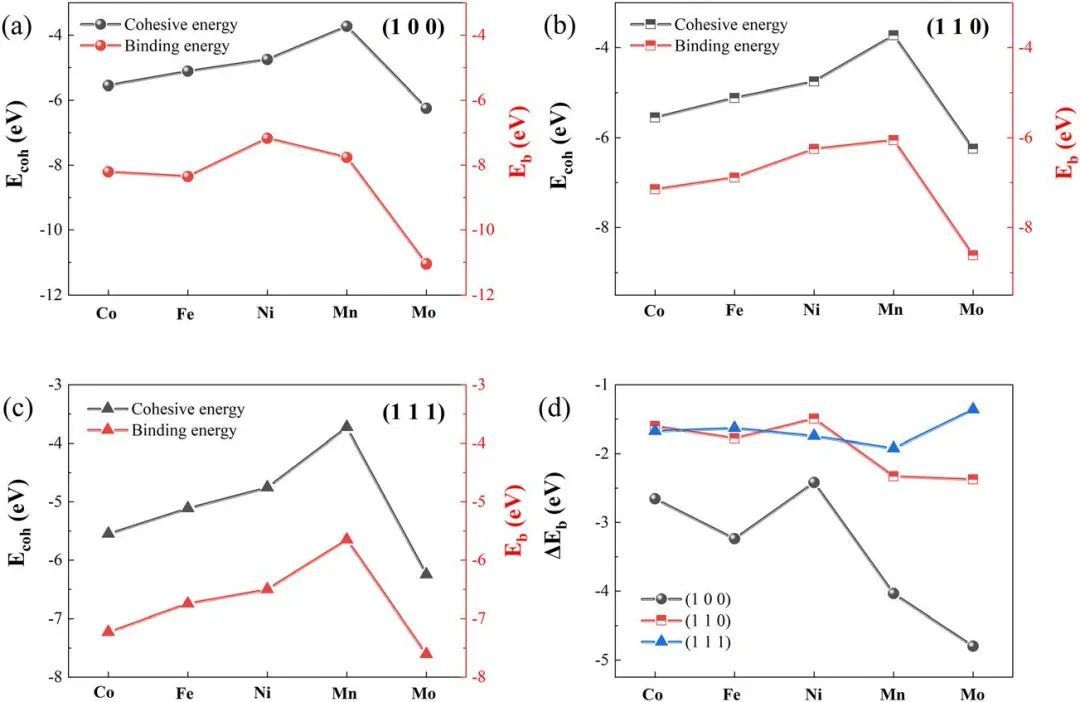
图2 催化剂的结合能与内聚能
为了验证TM原子的表面掺杂是否真的对VC的HER性能有积极的贡献,分别在(100)、(110)和(111)三个平面上计算了不同TM原子掺杂前后的ΔGH*。纯VC(100) C位点的ΔGH*为0.18013 eV,表明H原子在HER中俘获活性位点的能力较弱。Mn-VC(100)的C位ΔGH*值为0.00117 eV,远小于未掺杂VC,表现出最佳的HER性能,如图3(a)所示。这表明Mn元素的引入确实有利于增强H原子对模型表面的吸附力。
无论TM原子是否掺杂在(100)平面上,C位都是HER的主要活性位。而原始VC的桥式V1V2位点的ΔGH*为负,约为0.09504 eV,说明其表面对H原子具有较强的吸附能力,阻碍了H在(110)表面上的解离。此外,TM掺杂有效地调节了VC表面与H原子的相互作用,促进VC表面的HER动力学过程,如图3(b)所示。Co-VC的桥式V1Co位点催化活性最高,ΔGH*为0.014649 eV。Fe-VC的桥式V1Fe位点、Ni-VC的桥式V1V2位点、Mn-VC的桥式V1Mn位点和Mo-VC的桥式V1V2位点上的ΔGH*分别为0.02086、0.02151、0.01819和0.08730 eV。显然,对于HER, V原子和TM掺杂原子之间的桥活性位点在(110)平面上更活跃。图3(c)给出了TM掺杂VC(111)计算得到的ΔGH*。
由图可知,纯净VC的(111)平面上有较大的正ΔGH*(0.12530 eV),说明纯VC的V位对H原子的吸收能力较弱,这将阻碍HER的进行。Ni的掺杂适当地增强了VC对H原子的表面吸引力,Ni-VC(111) Ni位上的ΔGH*为0.03889 eV。此外,Co-VC、Fe-VC和Mn-VC的V位DGH*值与(111)平面分别为0.08011、0.08304、0.11802和0.1403753 eV。图3(d)展示了所有计算模型的火山图,直观地对比了它们的HER性能。越靠近火山图的顶点,ΔGH*值越接近0,交换电流密度越高,表明HER性能越好。HER表现较好的模型用虚线框圈出。
从三种不同暴露晶面下的TM掺杂VC模型的催化反应性能来看,Co、Fe、Ni和Mo掺杂VC模型在暴露晶面为(110)面时具有较高的催化反应活性,计算得到的VC(110)模型整体上具有较高的HER催化活性,Mn的掺杂可以有效提高(100)和(110)晶面上的HER性能。
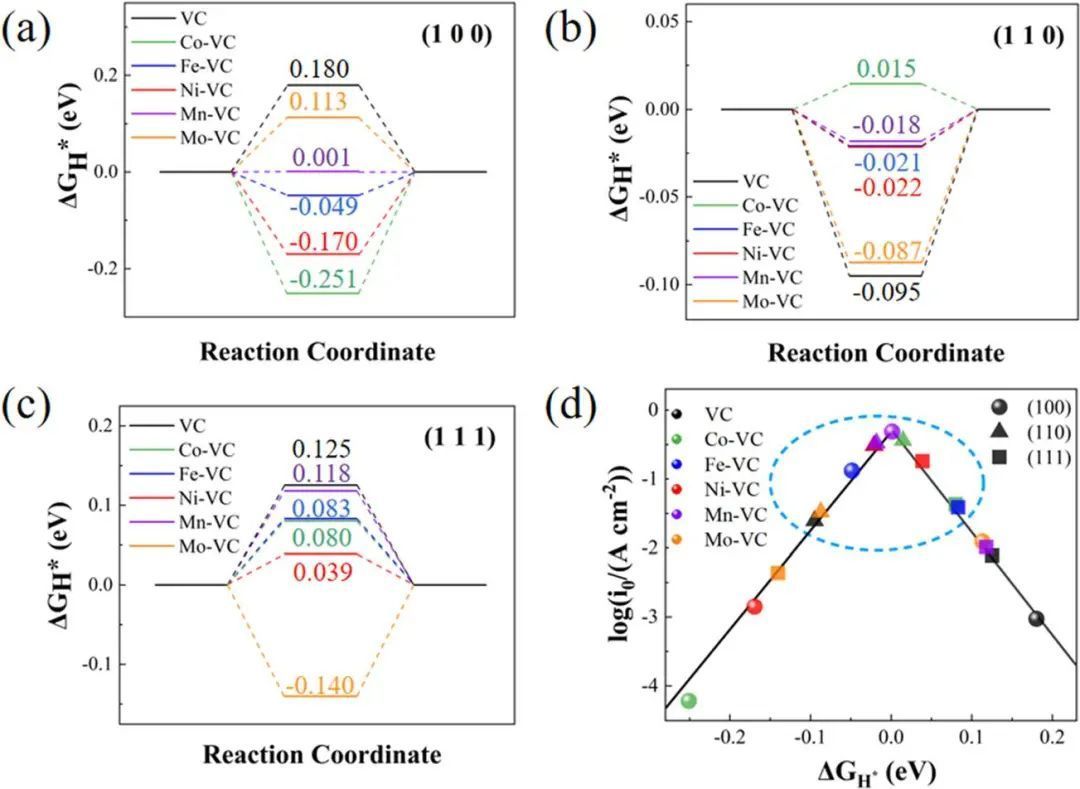
图3 催化剂的析氢自由能与火山曲线
值得注意的是,电催化剂优异的电子性能是实现高电催化性能的基础。然而,电催化剂的固有电子性质与其电子结构密切相关。计算了它们的电子态密度,探讨了掺杂原子对电子结构的调制作用,并探讨了提高HER性能的机理。如图4(a)所示,在VC(100)中掺杂Mn原子后,C 2p轨道在费米能级附近的投射电子态密度高于纯VC。调制后的电子结构有利于载流子的快速迁移,进而激活被吸附的H原子,参与后续的HER。
此外,如图4(b)所示,Co-VC模型费米能级附近的Co 3d轨道的投影电子态密度明显高于纯VC模型的V 3d轨道,并且在费米能级附近存在多个峰值。Co原子的引入有效地增加了费米能级附近的电子数,有利于提高VC在(110)平面上的电催化活性。
此外,如图4(c)所示,Ni-VC(111)的Ni 3d轨道的PDOS在费米能级附近较VC(111)的V 3d轨道有一个较高的峰值,这有助于潜在的电催化活性。综上所述,TM原子的掺杂成功地提高了VC在费米能级附近的电子态密度,调整了VC的电子结构。
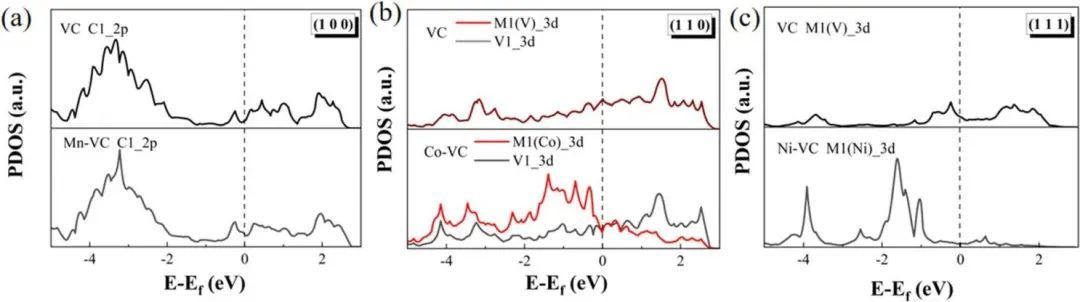
图4 催化剂态密度
金属3d轨道与分子轨道相互作用,导致分子轨道能级分裂,形成低成键轨道和高反键轨道。当反键轨道高于费米能级时,反键轨道不被电子回流所占据,有利于吸附。此外,随着d带中心的增大,吸附位点对应的ΔGH*也会降低。如图5所示,讨论了掺杂前后VC模型不同晶面上吸附位点附近原子d带中心与ΔGH*的线性关系。其中R为Pearson相关系数,用于解释不同VC模型中ΔGH*与d带中心的相关程度。
图5(a)为不同VC模型的(100)平面上V1吸附位点d带中心与ΔGH*的线性关系。VC(100)中V位的d-带中心为0.879 eV, ΔGH*为0.801 eV,说明V位的低d带中心导致H原子在(100)平面及更远处的吸附较弱,抑制VC的HER活性。掺杂Mo后,V1位点的d带中心显著增大(0.846 eV),使H吸附增强,从而提高了HER性能。
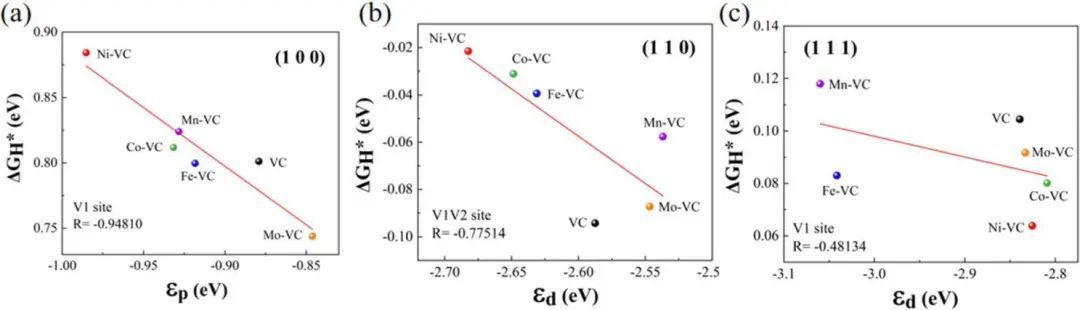
图5 催化剂析氢自由能与d带中心的标度关系
图6分别为VC模型和掺TM 的VC模型(100)、(110)和(111)面电荷密度差图。青色部分代表电子的损失,黄色部分代表电子的获得。对比图6(a)和(d)可以发现,在VC(100)中掺杂Mn原子有效地促进了电子从金属向非金属的转移。与Mn原子相邻的碳原子获得了更多的电子,导致负电荷的积累,使C位更容易吸收H原子。最终,表面吸附反应能垒的降低有利于HER过程。从图6(b)可以看出,纯VC的桥式V1V2位点失去更多电子,导致H原子吸附较强,G值为负。
而在图6(e)中,Co原子的掺杂促进了整个表面的电荷转移,缓解了局部的大量电子损失,导致H在金属位点上的吸附力下降。同样,在纯VC(111)模型中,青色部分表示V位电子损失较大,如图6(c)所示,表明其与H原子相互作用弱。然而,Ni的引入大大改善了VC(111)中的电荷转移行为,特别是图6(f)中Ni位点附近的电子损失显著降低。综上所述,活性位点附近的电荷转移对催化剂表面H吸附和解吸反应的能垒有重要影响。
通过掺杂过渡金属原子,可以调节VC不同晶面上活性位点附近的电荷分布和转移,从而更好地匹配H在这些表面的吸附和解吸过程,从而促进HER的催化反应动力学。
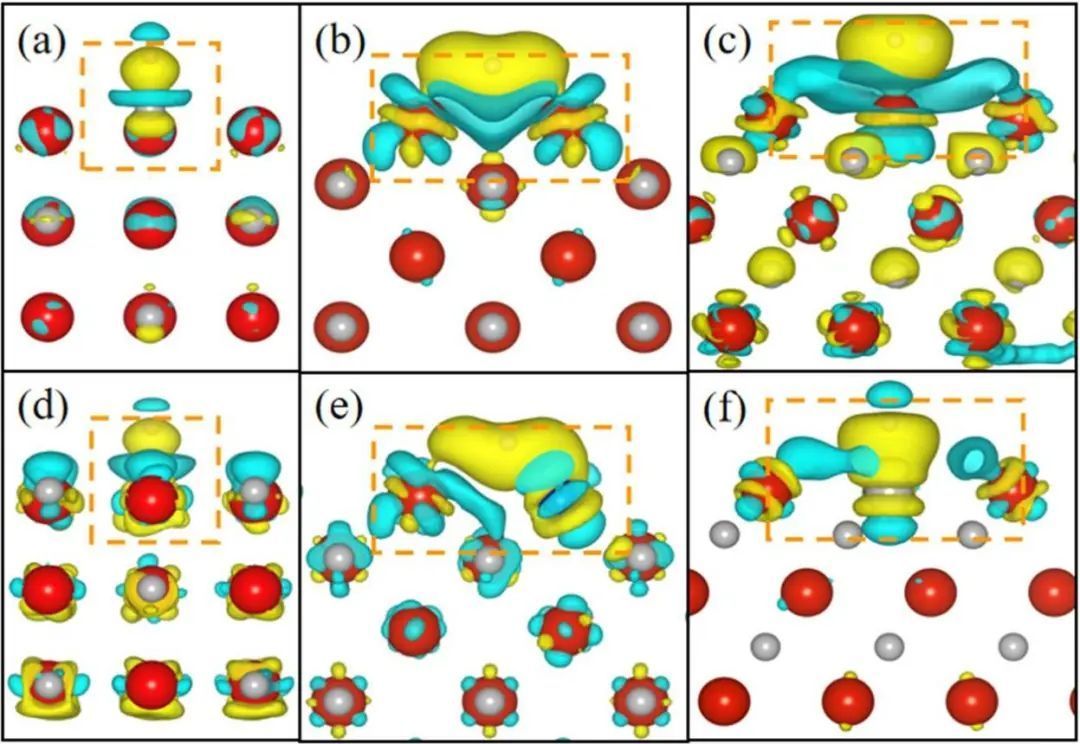
图6 催化剂差分电荷密度
随着pH值的增加,ΔGH*值也随之增加,ΔGH*值越接近0,HER性能越好。图7为pH在1 ~ 14范围内不同模型的ΔGH*值。如图7(a)所示,与TM掺杂VC相比,纯VC在所有pH值下都表现出较差的HER性能,而Co-VC(100)和Ni-VC(100)在pH值0 ~ 8范围内表现出较高的催化活性。其中Co-VC在pH = 4.5时ΔGH*接近于零,与其他模型相比,需要较弱的酸性环境。
从图7(b)可以看出,酸性越强的环境对VC(110)的HER性能越有利,在pH值约为0 ~ 4的范围内,其催化反应动力学越快。而在(110)平面上,相同pH条件下,不同TM掺杂对ΔGH*影响不大。原始VC(111)和掺杂Co、Fe、Ni、Mn的VC(111)模型在超强酸环境中表现出较高的催化活性。
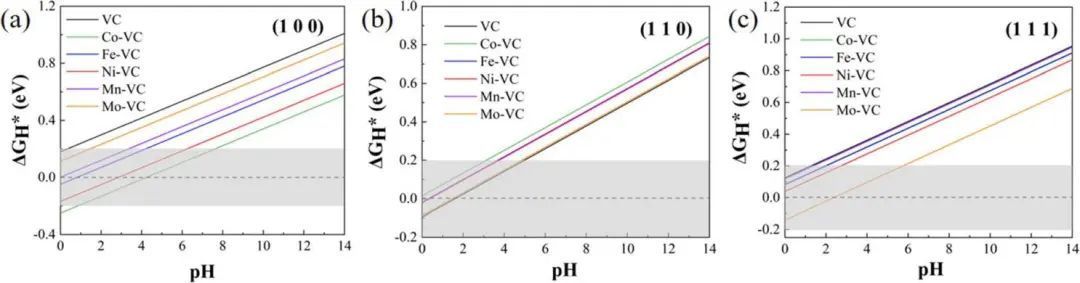
图7 催化剂析氢自由能与pH的变化关系
结论与展望
本文通过DFT计算对过渡金属(Co, Fe, Ni, Mn, Mo)掺杂VC用于电催化HER进行了全面的理论研究,其中在VC(100)中掺杂Co显著拓宽了高效HER所需的pH范围。这项工作揭示了掺杂是提高电催化析氢性能的有效途径,根据催化剂暴露的晶面选择合适的掺杂原子有利于在实际应用中设计高性能的HER催化剂。
文献信息
Zhang, Y., Zhang, B., Tong, L., Xing, J., & Fu, X. (2023). Computational screening toward transition metal doped vanadium carbides in different crystal planes for efficient hydrogen evolution: a first-principles study. Physical Chemistry Chemical Physics.ht-tps://doi.org/10.1039/D2CP05207E


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









